Animal Drug User Fee Programs
The Food and Drug Administration’s (FDA’s) review of brand-name and generic animal drug applications is funded through a combination of annual discretionary appropriations from Congress and user fees collected from the regulated industry.
The Animal Drug User Fee Act of 2003 (ADUFA I, P.L. 108-130) gave FDA initial authority to collect user fees from sponsors to improve the timeliness of review of animal drug applications. ADUFA I did not cover generic animal drugs. In 2008, in response to concerns regarding generic drug application review times and a backlog of applications, Congress passed legislation that reauthorized ADUFA and created a new user fee program for generic animal drugs. Title II of P.L. 110-316, the Animal Generic Drug User Fee Act (AGDUFA I), provided FDA with authority to collect user fees for the review of generic animal drug applications.
Under both its brand-name and generic animal drug authorities, FDA may use fee revenue only for the costs of “the process for the review” of the respective animal drug type, which includes the following: review of applications and other submissions; facility inspections; oversight of research to support the application/submission; issuance of regulations, policies, standards, and action letters; and review of labeling and advertising prior to approval. Neither authority permits postmarket review activities under current law.
Authority to collect user fees for animal drug review has been written to sunset at five-year intervals; as a result, reauthorization is often considered to be “must pass” legislation. Congress last reauthorized ADUFA and AGDUFA through September 30, 2018, via the Animal Drug and Animal Generic Drug User Fee Reauthorization Act of 2013 (P.L. 113-14). ADUFA and AGDUFA reauthorization consists of two parts: (1) statutory language that reauthorizes the programs, and (2) the negotiated agreement on performance goals and procedures between FDA and industry for the upcoming five-year interval. FDA is tasked by law with specific responsibilities in the reauthorization process.
As required for the upcoming reauthorization, in May 2016 FDA held two public meetings, one each for ADUFA and AGDUFA, to begin the process. For the next several months through early 2017, FDA held separate negotiations with the brand-name and generic animal drug industries, and met with other stakeholder groups (scientific and academic experts, veterinary professionals, patient and consumer advocacy groups). On October 25, 2017, FDA published in the Federal Register the “ADUFA IV” and “ADGUFA III” reauthorization proposals, including commitments negotiated with industry. On November 2, 2017, the agency held two public meetings, one each for ADUFA and AGDUFA, to discuss the proposed recommendations and commitments. Pursuant to the statute, these recommendations must be submitted to Congress by January 15, 2018.
Unlike user fee authorities which sunset every five years, provisions in law that address FDA’s regulation of animal drugs are typically permanent. They are not part of the statutory language that authorizes user fees, and they are not subject to sunset. However, ADUFA /AGDUFA reauthorization offers a rare, “must pass” legislative focus on animal drug issues. Amendments dealing with other animal drug issues of interest to Congress, such as the use of antimicrobial drugs in food-producing animals, are often considered during the reauthorization process.
Animal Drug User Fee Programs
Jump to Main Text of Report
Contents
- Background
- Introduction
- Review of Animal Drugs
- Brand-Name (Pioneer) Animal Drugs
- Generic Animal Drugs
- Drug Use in Food-Producing Animals
- User Fees and the FDA Budget
- Animal Drug User Fee Programs
- Brand-Name (Pioneer) Animal Drugs and ADUFA
- Types of Fees and Fee Waivers
- Fee "Triggers": Authority to Collect Fees
- Performance
- Reauthorization
- Proposed ADUFA IV
- Generic Animal Drugs and AGDUFA
- Types of Fees and Fee Waivers
- Fee "Triggers": Authority to Collect Fees
- Performance
- Reauthorization
- Proposed AGDUFA III
Tables
- Table 1. Statutory Fee Revenue Amounts for Brand-Name Animal Drug Review: FY2014-FY2018
- Table 2. ADUFA Final Performance for FY2015
- Table 3. Selected ADUFA III and Proposed ADUFA IV Performance Goals
- Table 4. Statutory Fee Revenue Amounts for Generic Animal Drug Review: FY2014-FY2018
- Table 5. AGDUFA I Performance Goals
- Table 6. AGDUFA Final Performance for FY2015
- Table 7. AGDUFA II and Proposed AGDUFA III Performance Goals
- Table A-1. Summary of FDA Animal Drug User Fee Programs
- Table C-1. Animal Drug Program Activity Data
Summary
The Food and Drug Administration's (FDA's) review of brand-name and generic animal drug applications is funded through a combination of annual discretionary appropriations from Congress and user fees collected from the regulated industry.
The Animal Drug User Fee Act of 2003 (ADUFA I, P.L. 108-130) gave FDA initial authority to collect user fees from sponsors to improve the timeliness of review of animal drug applications. ADUFA I did not cover generic animal drugs. In 2008, in response to concerns regarding generic drug application review times and a backlog of applications, Congress passed legislation that reauthorized ADUFA and created a new user fee program for generic animal drugs. Title II of P.L. 110-316, the Animal Generic Drug User Fee Act (AGDUFA I), provided FDA with authority to collect user fees for the review of generic animal drug applications.
Under both its brand-name and generic animal drug authorities, FDA may use fee revenue only for the costs of "the process for the review" of the respective animal drug type, which includes the following: review of applications and other submissions; facility inspections; oversight of research to support the application/submission; issuance of regulations, policies, standards, and action letters; and review of labeling and advertising prior to approval. Neither authority permits postmarket review activities under current law.
Authority to collect user fees for animal drug review has been written to sunset at five-year intervals; as a result, reauthorization is often considered to be "must pass" legislation. Congress last reauthorized ADUFA and AGDUFA through September 30, 2018, via the Animal Drug and Animal Generic Drug User Fee Reauthorization Act of 2013 (P.L. 113-14). ADUFA and AGDUFA reauthorization consists of two parts: (1) statutory language that reauthorizes the programs, and (2) the negotiated agreement on performance goals and procedures between FDA and industry for the upcoming five-year interval. FDA is tasked by law with specific responsibilities in the reauthorization process.
As required for the upcoming reauthorization, in May 2016 FDA held two public meetings, one each for ADUFA and AGDUFA, to begin the process. For the next several months through early 2017, FDA held separate negotiations with the brand-name and generic animal drug industries, and met with other stakeholder groups (scientific and academic experts, veterinary professionals, patient and consumer advocacy groups). On October 25, 2017, FDA published in the Federal Register the "ADUFA IV" and "ADGUFA III" reauthorization proposals, including commitments negotiated with industry. On November 2, 2017, the agency held two public meetings, one each for ADUFA and AGDUFA, to discuss the proposed recommendations and commitments. Pursuant to the statute, these recommendations must be submitted to Congress by January 15, 2018.
Unlike user fee authorities which sunset every five years, provisions in law that address FDA's regulation of animal drugs are typically permanent. They are not part of the statutory language that authorizes user fees, and they are not subject to sunset. However, ADUFA /AGDUFA reauthorization offers a rare, "must pass" legislative focus on animal drug issues. Amendments dealing with other animal drug issues of interest to Congress, such as the use of antimicrobial drugs in food-producing animals, are often considered during the reauthorization process.
Background
Introduction
The Federal Food, Drug, and Cosmetic Act (FFDCA) authorizes the Food and Drug Administration (FDA) to regulate the safety and effectiveness of animal drugs. FDA's review of brand-name and generic animal drug applications is funded through a combination of annual discretionary appropriations from Congress and user fees collected from the regulated industry.
Congress first authorized FDA's collection of user fees for brand-name animal drugs for FY2004 in the Animal Drug User Fee Act of 2003 (now called ADUFA I, P.L. 108-130), generally following the model established for human drugs a decade earlier.1 At that time, representatives of brand-name animal drug research and development companies said that an animal drug could take 7 to 10 years to develop, at a cost of $100 million or more, and that review of these products had not always been timely or predictable.2
Authority to collect user fees for brand-name (pioneer) animal drugs has been written to sunset at five-year intervals, and was reauthorized in 2008 and 2013 in laws referred to as ADUFA II and ADUFA III, respectively. This authority is due to sunset at the end of FY2018. As required by law, FDA has published the results of negotiations with animal drug sponsors, which it refers to as ADUFA IV.3
Congress first authorized FDA's collection of user fees for generic animal drugs for FY2009 in the Animal Generic Drug User Fee Act of 2008 (AGDUFA I, P.L. 110-316), coincident with ADUFA II. AGDUFA was reauthorized in 2013 as AGDUFA II, and, like ADUFA III, is also due to sunset at the end of FY2018. As required by law, FDA has published the results of negotiations with generic animal drug sponsors, which it refers to as AGDUFA III.4
Appendix A provides a snapshot of the legislative histories, statutory citations, and key statistics for both user fee programs.
Under both its brand-name and generic animal drug authorities, FDA shall use fee revenue for the costs of "the process for the review" of the respective animal drug type, which includes the following: review of applications and other submissions; facility inspections; oversight of research to support the application/submission; issuance of regulations, policies, standards, and action letters; and review of labeling and advertising prior to approval.5 Neither authority permits postmarket review activities under current law.
Sunset provisions, which are found in other FDA user fee authorities (such as those for human drugs and devices), may set a more strict reauthorization timeline for Congress and the agency than do the more common expirations of authority for appropriations. Despite a lapsed authorization of appropriations, HHS agencies may invoke general authorities to continue their work on a matter as the authority to conduct the activity remains. A sunset, in contrast, bars the agency from continuing such work beyond the sunset date, unless or until Congress reauthorizes the provision or otherwise lifts the sunset.
The key consequence for FDA and drug sponsors of a lapse in user fee authority is that continued payment of FDA review personnel is prohibited, and the agency may have to lay off animal drug review staff. These personnel are highly specialized and not easily replaced. As a result, user fee reauthorizations are often considered "must pass," and Congress has consistently reauthorized ADUFA and AGDUFA before their sunset dates.
Review of Animal Drugs
Brand-Name (Pioneer) Animal Drugs
|
Brand-name Animal Drug Submission Types: NADA – New Animal Drug Application, including all amendments and supplements INAD – Investigational New Animal Drug QLS – Qualifying Labeling Supplements |
Authorities governing FDA's review of animal drugs are found in FFDCA Section 512, which cross-references additional sections of the Act. Regulations are at 21 C.F.R. Chapter I, Subchapter E, which also cross-references other regulations. FDA does not oversee veterinary biologics (e.g., vaccines), which are regulated by the Department of Agriculture's Animal and Plant Health Inspection Service (APHIS). The FDA's Office of New Animal Drug Evaluation (ONADE) determines whether or not a new animal drug application (NADA) should be approved for marketing. According to ONADE, in order to approve and sustain a new animal drug for commercial use, five standards must be met.
- 1. The drug must be safe for the animal, for humans consuming food derived from a treated food-producing animal, and for the person administering the drug.
- 2. The drug must be effective for its intended uses, i.e., the uses prescribed, recommended or suggested in the labeling of the product.
- 3. The drug must be a quality manufactured product, being the result of a validated manufacturing process conducted in accordance with current Good Manufacturing Practice (GMP) regulations.
- 4. The drug must be properly labeled to inform the user of the product not only how to use the product but also safety considerations, procedures for use in food-producing animals, and procedures for storage and handling.6
- 5. Under the National Environmental Policy Act, FDA must consider, during the review process, the environmental impact of an animal drug's postmarket use (e.g., medicated feed waste and potential runoff).7
In 2004 Congress addressed concerns about the availability of some animal drugs that are not commercially attractive, and enacted the Minor Use and Minor Species Animal Health Act (MUMS, P.L. 108-282), modeled after the authority for "orphan" human drugs. A minor use is a use in a major species (horses, dogs, cats, cattle, pigs, turkeys, and chickens) for a disease that occurs infrequently, involving only a small number of animals annually. Minor species are all other nonhuman animal species (except for the above listed major species)—such as zoo animals, fish, and pet birds and rodents—including some species of food-producing animals—such as sheep, goats, catfish, game birds, and honey bees, among others. MUMS allows conditional approval (marketing before all effectiveness data are available) and a period of marketing exclusivity, during which FDA may not approve an application for the generic copy of the approved brand-name drug.8 FDA may waive user fee requirements under ADUFA and AGDUFA for MUMS drugs.
As with human medical products, it is the responsibility of the drug sponsor to conduct the necessary tests to demonstrate a product's safety and effectiveness for review purposes. Once the drug is on the market, FDA continues to monitor the animal drug's safety and effectiveness, manufacturing processes, labeling, and marketing communications.9
For the basic types of applications and other submissions in the review of a brand-name animal drug, as well as commonly used acronyms, see text box, above.
Generic Animal Drugs
|
Generic Animal Drug Submissions and References: ANADA – Abbreviated New Animal Drug Application, including all amendments and supplements JINAD – Generic Investigational New Animal Drug RLNAD — Reference-Listed New Animal Drug, the brand drug to which the generic is compared |
Like brand-name animal drugs, authorities governing FDA's review of generic animal drugs are found in FFDCA Section 512, and regulations are at 21 C.F.R. Chapter I, Subchapter E. The Generic Animal Drug and Patent Term Restoration Act of 1988 (P.L. 100-670) created an expedited pathway for the review of generic animal drugs. This pathway allows a sponsor to submit an abbreviated new animal drug application (ANADA) to FDA for premarket review rather than the full NADA required for a brand-name animal drug. The process is called "abbreviated" because instead of conducting new safety and effectiveness testing, the ANADA sponsor establishes that the generic animal drug is the same as the reference-listed new animal drug product (RLNAD, which is the brand drug), thereby relying on FDA's determination that the brand drug is safe and effective. In the ANADA, the generic drug sponsor must show: that the generic copy has the same active ingredient, strength, dosage form, dosing regimen, and route of administration as the RLNAD; that it is bioequivalent (or that the bioequivalence requirement has been waived); and that it is the same in identity, strength, purity, and quality.10 The labeling for the generic animal drug also must be the same as the brand, with some exceptions (e.g., company name and address). An ANADA also must contain an environmental assessment (EA) or a request for categorical exclusion from the EA, among other information.11
For basic types of applications, other submissions, and product references in the review of a generic animal drug, as well as commonly used acronyms, see text box, above.
Drug Use in Food-Producing Animals
As noted, FDA must consider public health and environmental impacts in its safety assessment of a proposed new animal drug. This is most evident in FDA's regulation of drugs used in food-producing animals, where use of drugs can affect the safety of meat and milk products consumed by humans or other animals. FDA applies the following approaches, among others, to the review, approval, and use of these drugs.
- Tolerances and Withdrawal/Withholding/Discard Periods. Drug sponsors are required to study the persistence of a drug (called a "residue") in the tissues of food-producing animals and demonstrate a safe management approach, such as the minimum number of days after drug treatment ends that an animal must be held untreated before it can go to slaughter (called a "withdrawal period"), or that a cow's milk must be discarded rather than marketed.12
- Limitations on "Extra-Label" Prescribing. Veterinarians are generally permitted to prescribe and/or dispense medications for indications not stated on the label (referred to as "off-label" in human medicine) and for species not stated on the label. This provides the basis of authority for veterinarians to prescribe or dispense human medications for use in animals. However, this practice is limited in a number of ways for uses in food-producing animals, in order to keep the food supply free of potentially harmful drug residues.13
- Judicious Use of Medically Important Antimicrobial Drugs. According to FDA, "Antimicrobial use in animals can contribute to the emergence of antimicrobial resistance in bacteria that may be transferred to humans, thereby reducing the effectiveness of antimicrobial drugs for treating human disease."14 The agency has focused attention in particular on food-producing animals, which can transmit foodborne infections in their meat or milk, and on what it calls "production uses" of antimicrobial drugs, intended to promote weight gain or growth efficiency rather than as treatment for specific diseases. In 2010, FDA published draft guidance (which it finalized in 2012) stating principles for limiting medically important antimicrobial drugs to uses in food-producing animals that are considered necessary for assuring animal health, and limiting such drugs to uses in food-producing animals that include veterinary oversight or consultation.15 This effectively limited new approvals to drugs for which there was evidence of effectiveness against a specific disease or condition, and eliminated new approvals for over-the-counter antimicrobial uses. In 2013, FDA asked sponsors of already approved antimicrobial drugs used in food-producing animals to either demonstrate evidence of effectiveness for a disease treatment or prevention indication, or to voluntarily withdraw the drug.16 In January 2017, FDA announced the completion of this process, which resulted in 84 voluntary product withdrawals and enhanced controls over the ongoing use of remaining products in food-producing animals.17
- Regulation of Feed Mills. Food-producing animals are often fed blended feed mixtures containing vitamins and minerals, to which drugs are also sometimes added. These blends are produced commercially by feed mills, which came under increased scrutiny pursuant to the Animal Drug Availability Act of 1996 (P.L. 104-250). In 2015, through rulemaking, FDA expanded the role of veterinarians in the use of antimicrobial drugs in blended feeds (called the "Veterinary Feed Directive" or VFD) in order to assure the safe use of these drugs in food-producing animals.18
- Reporting of Antimicrobial Animal Drug Sales and Distribution. Section 105 of ADUFA II requires that drug sponsors annually report to FDA the amount of antimicrobial drugs they sell or distribute for use in food-producing animals, and requires FDA to issue annual summary reports of the sales and distribution data.19 Sponsors have said that they are not in a position to know how products are actually used once distributed (e.g., the species or condition being treated), although such usage information is potentially valuable in monitoring and addressing concerns about antimicrobial resistance. In 2016, through rulemaking, FDA began requiring sponsors to provide FDA with estimates of sales broken out by major food-producing species (cattle, swine, chickens, and turkeys).20
Authorities that address FDA's regulation of animal drugs used in food-producing animals are generally permanent. They are not part of the statutory language in FFDCA Title VII that authorizes user fees and is subject to sunset. However, ADUFA /AGDUFA reauthorization offers a rare legislative focus on animal drug issues, and amendments dealing with these and other aspects of animal drug use are often considered by Congress during the reauthorization process.
User Fees and the FDA Budget
FDA's budget has two funding streams: annual appropriations (i.e., discretionary budget authority) and industry user fees. In FDA's annual appropriation, Congress sets both the total amount of appropriated funds and the amount of user fees that the agency is authorized to collect and obligate for that fiscal year. FDA's program level (i.e., total budget) in FY2016 was $4.745 billion, with user fees accounting for 43% of the enacted program level.21
FDA's Center for Veterinary Medicine (CVM) is responsible for ensuring the safety and effectiveness of brand and generic animal drugs.22 FDA financial reports, required by statute, provide the total amount spent on the ADUFA and AGDUFA programs, including the dollar amount and percentage derived from user fees and nonuser fee appropriations. While most of ADUFA and AGDUFA revenue supports activities managed by CVM, revenue from both user fee programs also contributes to other FDA organizational components that support the ADUFA and AGDUFA programs, including the Office of Regulatory Affairs (ORA) and FDA headquarters. CVM incurs the cost for the review of application and investigational submissions; ORA incurs field inspection, investigation, and laboratory costs; and FDA headquarters incurs general and administrative costs.23
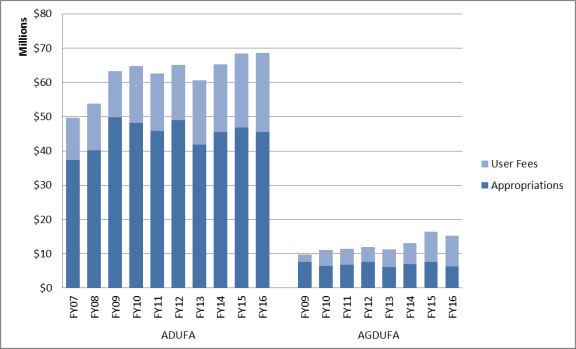
Animal drug user fees were first collected in FY2004 and since then, have generally comprised an increasing proportion of the FDA's budget that is focused on animal drug review. In FY2005, animal drug user fees provided 20% of the ADUFA program total costs (appropriations covered 80%).24 In FY2016, user fees provided 34% of ADUFA total costs (appropriations covered 66%).25
Generic animal drug user fees were first collected in FY2009 and also have generally comprised an increasing proportion of the FDA's budget that is focused on generic animal drug submissions. In FY2009, generic animal drug user fees comprised 22% of the AGDUFA program total costs (appropriations covered 78%). In FY2016, user fees comprised 59% of AGDUFA total costs (appropriations covered 41%).26
Appendix B provides information on the relative proportion of costs supported by user fee revenue and appropriations for the ADUFA and AGDUFA programs.
Animal Drug User Fee Programs
Brand-Name (Pioneer) Animal Drugs and ADUFA
Congress first authorized user fees for brand-name animal drug review in 2003 in response to the same concerns from animal drug sponsors that had spurred establishment of a user fee program for human drugs a decade earlier. Given the years of research and cost involved in animal drug development, sponsors argued for these fees to counter increasing review backlogs, which delayed market entry and therefore—they believed—compromised both animal health and future drug development.27
Types of Fees and Fee Waivers
Current law authorizes the collection of four types of user fees for brand-name animal drugs, and sets the total amount of revenue for each fee type for each covered fiscal year, to be adjusted annually for inflation and workload, as specified. ADUFA III provided for a final year adjustment, allowing the Secretary to increase fee amounts to provide for up to three months of operating reserves of carryover user fees for reviewing NADAs in the first three months of FY2019, unless FDA has carryover balances in excess of three months of such operating reserves. The four types of brand-name animal drug user fees are as follows.
- Application and supplement fee. A one-time fee for each NADA or supplement a sponsor submits (20% of estimated revenue).28
- Product fee. An annual fee for each new animal drug product a sponsor has submitted an application for, including approved and pending NADAs and supplemental applications (27% of estimated revenue).
- Establishment fee. An annual fee for each establishment that manufactures an animal drug product named in an approved or pending NADA or supplemental application during the given fiscal year (26% of estimated revenue).
- Sponsor fee. An annual fee for each sponsor of an approved or pending NADA, supplemental application, or INAD submission (27% of estimated revenue).
The statutory fee revenue amounts for FY2014-FY2018, as established by ADUFA III, are listed in Table 1.
Table 1. Statutory Fee Revenue Amounts for Brand-Name Animal Drug Review:
FY2014-FY2018
Dollars in thousands
|
Type of fee |
FY2014 |
FY2015 |
FY2016 |
FY2017 |
FY2018 |
TOTAL |
|
Application |
4,720 |
4,320 |
4,320 |
4,320 |
4,320 |
22,000 |
|
Product |
6,372 |
5,832 |
5,832 |
5,832 |
5,832 |
29,700 |
|
Establishment |
6,136 |
5,616 |
5,616 |
5,616 |
5,616 |
28,600 |
|
Sponsor |
6,372 |
5,832 |
5,832 |
5,832 |
5,832 |
29,700 |
|
TOTAL |
23,600 |
21,600 |
21,600 |
21,600 |
21,600 |
110,000 |
Source: Calculated by Congressional Research Service from FFDCA §740(b).
Notes: Amounts do not reflect adjustments for inflation or review workload.
The law requires the Secretary to set and publish in the Federal Register annual fees, based on the statutory revenue amounts and adjustments, 60 days before the start of each fiscal year. The Secretary may waive or reduce fees if he or she determines (1) assessment of the fee would present a significant barrier to innovation; (2) the fees to be paid will exceed expected review costs; (3) the animal drug is intended to be used in a medicated feed;29 (4) the animal drug is intended solely for a MUMS indication; or (5) the sponsor is a small business (defined as having fewer than 500 employees) submitting its first animal drug application. Information about the numbers and value of waivers and reductions granted and used is provided in FDA's annual ADUFA financial reports.30
Fee "Triggers": Authority to Collect Fees
To ensure that user fees supplement, rather than replace, appropriated funds, the law includes three funding "triggers" that prohibit FDA from collecting user fees for animal drug review unless certain conditions are met each year. FDA may collect and use fees only if the nonuser fee appropriations, both for the activities involved in the review of animal drug applications and for FDA activities overall, remain at a level at least equal (adjusted for inflation) to FY2003.31 The third trigger prohibits FDA from collecting user fees in excess of the amount specified in annual appropriations or otherwise made available for obligation that fiscal year.32
Performance
ADUFA requires the Secretary to submit to Congress, not later than 120 days before the end of each fiscal year for which fees are collected, a performance report concerning FDA's progress in achieving the performance goals identified in the ADUFA agreement letter, and a financial report on the implementation of the user fee authority and the use of fees collected during that fiscal year.33
In FY2011 FDA released an electronic tool for animal drug review submissions (called eSubmitter), in order to improve review efficiency. In FY2015 and FY2016, about 70% of all submissions were made electronically.34
According to FDA's most recent performance report (for FY2016), the agency exceeded its ADUFA III performance goals for FY2015 (as shown in Table 2), and for the first three quarters of FY2016.35
|
Submission Type |
Review Time Goal: Act on 90% within:a |
# Filedb |
# Reviewed on Time |
# Overdue |
% on Time |
|
Original NADAs and reactivations |
180 days |
3 |
3 |
0 |
100% |
|
Administrative NADAs |
60 days |
16 |
16 |
0 |
100% |
|
Nonmanufacturing supplemental NADAs and reactivations |
180 days |
6 |
6 |
0 |
100% |
|
Manufacturing supplemental NADAs and reactivations |
120 days |
327 |
327 |
0 |
100% |
|
Qualifying labeling supplements |
60 days |
3 |
3 |
0 |
100% |
|
INAD studies |
180 days |
147 |
147 |
0 |
100% |
|
INAD study protocols |
60 days |
248 |
246 |
2 |
99% |
Source: Adapted by Congressional Research Service from FDA, "FY2016 Performance Report to Congress for the Animal Drug User Fee Act," p. 7, undated, https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/ucm537556.htm.
Notes: For determining performance goals, FDA defines supplemental applications that are "major" (most supplemental NADAs), "moderate," or "minor," depending on how likely the change is to have an adverse effect on the identity, strength, quality, purity, or potency of the drug. A nonmanufacturing supplemental is considered a major change. A manufacturing supplemental is limited to a manufacturing change necessitated by an unforeseen event (such as a fire), for which expedited review is provided. FDA, "Guidance for Industry: Chemistry, Manufacturing, and Controls Changes to an Approved NADA or ANADA," p. 6, May 30, 2007.
a. FDA defines "act on" to mean that it completes review of a submission and issues a final letter to the sponsor either stating approval (or, for an INAD, completeness of the submission), or setting forth the deficiencies that need to be addressed as a condition of approval.
b. Excludes filings that were found to be insufficient on their face or that were withdrawn.
Reauthorization
FFDCA Section 740A(d), as amended by ADUFA III, sets forth the process for reauthorization, directing FDA to develop recommendations for the following five fiscal years (i.e., FY2019-FY2023) in consultation with specified congressional committees, scientific and academic experts, veterinary professionals, patient and consumer advocacy groups, and the regulated industry. Prior to negotiations with industry, FDA is required to request public input, hold a public meeting, provide a 30-day comment period, and publish public comments on the agency's website. During negotiations with industry, not less frequently than once every four months, FDA must hold discussions with representatives of veterinary, patient, and consumer advocacy groups to receive their suggestions and discuss their views on the reauthorization. After negotiations with industry are completed, FDA is required to present the recommendations to the specified congressional committees, publish the recommendations in the Federal Register, provide a 30-day public comment period, hold another public meeting to receive views from stakeholders, and revise the recommendations as necessary. Minutes of all negotiation meetings between FDA and industry must be posted on the FDA website.36
Proposed ADUFA IV
FDA has published on its website a summary of the ADUFA IV proposed statutory changes, as well as a notice in the Federal Register announcing the availability of the ADUFA IV draft recommendations.37 According to the agency's summary, the proposed ADUFA IV agreement with industry would:
- require sponsors to submit all applications and other submissions electronically, beginning October 1, 2018;
- retain all performance goals from ADUFA III, with the exception of manufacturing supplemental NADAs and supplements with changes being effected, for which a longer review time was agreed upon (See Table 3); and
- add performance goals for review of certain medicated feed applications, for conduct of presubmission conferences, and for pilot studies on new test methods to detect drug residues in tissues of food-producing animals.38
The draft ADUFA IV agreement also proposes several changes to ADUFA III authority, specifically to:
- continue the inflation adjustment, and the workload adjustment, as calculated per CVM policy, except to reset the base years to FY2014 through FY2018, with no workload adjustment for FY2019. The proposed FY2019 baseline for ADUFA IV is $30,331,240, which includes a $400,000 one-time cost for information technology enhancement. For FY2020- FY2023, annual statutory revenue amounts would be further adjusted for inflation and workload, and would include $900,000 per year for research on tissue testing methods;
- eliminate the final year offset provision and make any excess collections available to "enhance the review process in real time;" and
- authorize the Secretary, when setting fees, to reduce a calculated workload adjustment, as specified.
|
Submission Type |
Performance goal (in days): Act on 90% within: |
|||
|
ADUFA III |
ADUFA IV |
|||
|
Original NADAs and Reactivations |
|
|
||
|
Administrative NADAs |
|
|
||
|
Nonmanufacturing Supplemental NADAs and Reactivations |
|
|
||
|
Manufacturing Supplemental NADAs and Reactivations |
|
|
||
|
Manufacturing Supplemental NADAs and Reactivations |
|
|
||
|
Qualifying Labeling Supplements |
|
|
||
|
Investigational New Animal Drug (INAD) Studies |
|
|
||
|
INAD Study Protocols |
|
|
||
Source: 82 Federal Register 49380, October 25, 2017; and FDA Presentation, "Animal Drug User Fee Act Reauthorization (ADUFA IV), FY 2019 –FY 2023" Public Meeting, November 2017, https://www.fda.gov/downloads/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/UCM585895.pdf.
Note: Supplementals for which changes are being effected are considered moderate changes that can be made by the sponsor while FDA review is pending.
The total five-year revenue planned for ADUFA III was $114 million. It is estimated that the total five-year revenue for ADUFA IV would be $150 million. The fee revenue distribution in ADUFA IV would remain the same as ADUFA III: 20% from application fees; 27% from product fees; 26% from establishment fees; and 27% from sponsor fees.
According to the agency's section by section summary of the proposed statutory changes, the proposed ADUFA IV, in addition to codifying several of the proposals in the draft agreement, would amend (1) the definition of "animal drug application" to include an application for conditional approval under FFDCA Section 571,39 and (2) the definition of "process for the review of animal drug applications" to include certain activities related to implementation of the US-European Union GMP Mutual Inspection Agreement.40 The proposed statutory language also would add an exemption from fees for certain labeling supplements (to add the number of the approved application to the labeling).41
Generic Animal Drugs and AGDUFA
FFDCA Section 512(c)(1) requires FDA to review and act on ANADAs within 180 days of submission.42 In congressional testimony in June 2008, FDA reported that in FY2007, the average review time for ANADAs was 570 days, and there was a backlog of 446 of these submissions, almost double the number in FY2000.43 At that time, the review of ANADAs was funded entirely through appropriations, and ADUFA user fee funds were not authorized to be applied to generic animal drug reviews. In 2008, Congress passed legislation authorizing FDA to assess and collect fees from generic animal drug sponsors to support the review of ANADAs, supplemental applications,44 and investigational submissions (i.e., generic investigational new animal drug [JINAD] submissions).45 In exchange for the authority to collect user fees, AGDUFA requires FDA to pursue certain performance goals negotiated between the agency and industry.
Types of Fees and Fee Waivers
Current law authorizes the collection of three types of user fees, and sets the total amount of revenue for each fee type for each covered fiscal year, to be adjusted annually for workload. The fee amounts established by AGDUFA I (for FY2009-FY2013) and AGDUFA II (for FY2014 through FY2018) included a fixed 4% inflation adjustment, and provided for a final year adjustment, allowing the Secretary to increase fee amounts to provide for up to three months of operating reserves of carryover user fees for reviewing ANADAs in the first three months of FY2019, unless FDA has carryover balances in excess of three months of such operating reserves. The three types of generic animal drug user fees are as follows.
- Abbreviated application fee. A one-time fee for each ANADA a sponsor submits (25% of estimated revenue).46
- Product fee. An annual fee for each generic new animal drug product a sponsor has submitted an application for, including approved and pending ANADAs and supplemental applications (37.5% of estimated revenue).
- Sponsor fee. An annual fee for each sponsor of an approved or pending ANADA, supplemental application, or investigational submission (37.5% of estimated revenue).47
The statutory fee revenue amounts for FY2014-FY2018, as established by AGDUFA II, are listed in Table 4.
Table 4. Statutory Fee Revenue Amounts for Generic Animal Drug Review:
FY2014-FY2018
(dollars in thousands)
|
Type of Fee |
FY2014 |
FY2015 |
FY2016 |
FY2017 |
FY2018 |
Total |
|
Application fees |
1,832 |
1,736 |
1,857 |
1,984 |
2,117 |
9,526 |
|
Product fees |
2,748 |
2,604 |
2,786 |
2,976 |
3,175 |
14,289 |
|
Sponsor fees |
2,748 |
2,604 |
2,786 |
2,976 |
3,175 |
14,289 |
|
TOTAL |
7,328 |
6,944 |
7,429 |
7,936 |
8,467 |
38,104 |
Source: FFDCA §741(b).
Notes: Amounts do not reflect adjustments for inflation or review workload.
The law requires the Secretary to set and publish in the Federal Register annual fees, based on the statutory revenue amounts and adjustments, at least 60 days before the start of each fiscal year. The Secretary may waive or reduce fees if he or she determines that the generic new animal drug is intended solely to provide for a MUMS indication.48 Information about the numbers and value of waivers and reductions granted and used is provided in FDA's annual AGDUFA financial reports.49
Fee "Triggers": Authority to Collect Fees
To ensure that user fees supplement, rather than replace, appropriated funds, the law includes three funding "triggers" (similar to the triggers for the ADUFA fees) that prohibit FDA from collecting user fees for generic animal drug review unless certain conditions are met each year. FDA may collect and use fees only if the nonuser fee appropriations for FDA activities overall remain at a level at least equal (adjusted for inflation) to FY2003, and if the nonuser fee appropriations for activities involved in the review of generic animal drug applications remain at a level at least equal (adjusted for inflation) to FY2008.50 The third trigger prohibits FDA from collecting user fees in excess of the amount specified in annual appropriations or otherwise made available for obligation that fiscal year.51
Performance
AGDUFA requires the Secretary to submit to Congress, not later than 120 days before the end of each fiscal year for which fees are collected, a performance report concerning FDA's progress in achieving the performance goals identified in the AGDUFA agreement letter and a financial report on the implementation of the user fee authority and the use of the fees collected during that fiscal year. AGDUFA I eliminated a backlog of 680 submissions, reduced submission review times from 700 days to 270 days, and met performance goals for all years with the exception of one goal (for JINAD protocols) in 2009.52 AGDUFA I phased in more challenging review goals over a five-year period (see Table 5).
|
Submission Type |
Performance goal (in days): Act on 90% within: |
||||
|
FY2009 |
FY2010 |
FY2011 |
FY2012 |
FY2013 |
|
|
Original ANADAs and Reactivations |
700 |
680 |
500 |
380 |
270 |
|
Administrative ANADAs |
120 |
115 |
110 |
105 |
100 |
|
Manufacturing Supplemental ANADAs and Reactivations |
600 |
570 |
420 |
270 |
270 |
|
JINAD Studies |
700 |
680 |
500 |
380 |
270 |
|
JINAD Protocols |
400 |
390 |
290 |
190 |
100 |
Source: FDA, Animal Generic Drug User Fee Act Performance Goals and Procedures, https://www.fda.gov/ForIndustry/UserFees/AnimalGenericDrugUserFeeActAGDUFA/ucm181567.htm.
Notes: An original application ANADA refers to an application that contains all the data for review as part of a complete application. An administrative application ANADA refers to an original or supplemental application for which data supporting discrete technical sections is submitted during the investigational phase of new animal drug review, allowing for "phased review" of the data. A JINAD study refers to a submission that contains both the data and conclusions of a study to be considered in support of the approval of a generic new animal drug. A JINAD protocol refers to a submission consisting of a protocol(s) or plan for conducting a study without substantial data.
AGDUFA II continued the review performance goals from FY2013, but added flexibility with a shortened review-time process for certain submissions, developed electronic submission capability, added a preapproval foreign inspection goal, and developed a question-based review for bioequivalence, among other things.53 According to FDA's most recent performance report (for FY2016), the agency exceeded its AGDUFA II performance goals for FY2015 (as shown in Table 6) and was on track to exceed all performance goals for FY2016.
|
Submission Type |
Review Time Goal: Act on 90% within: |
# Filed |
# Reviewed on Time |
# Overdue |
% on Time |
|
Original ANADAs and Reactivations |
270 days |
22 |
22 |
0 |
100% |
|
Administrative ANADAs |
100 days |
1 |
1 |
0 |
100% |
|
Manufacturing Supplemental ANADAs and Reactivations |
270 days |
152 |
151 |
1 |
99% |
|
JINAD Studies |
270 days |
54 |
53 |
1 |
98% |
|
JINAD Protocols |
100 days |
12 |
12 |
0 |
100% |
Source: Adapted by Congressional Research Service from FDA, "FY2016 Performance Report to Congress for the Animal Generic Drug User Fee Act," p. 13, undated, https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/UCM537571.pdf.
Reauthorization
FFDCA Section 742(d), as amended by AGDUFA II, sets forth the process for reauthorization of AGDUFA II, directing FDA to develop recommendations for the following five fiscal years in consultation with specified congressional committees, scientific and academic experts, veterinary professionals, patient and consumer advocacy groups, and the regulated industry. Prior to negotiations with industry, FDA is required to request public input, hold a public meeting, provide a 30-day comment period, and publish public comments on the agency's website. During negotiations with industry, not less frequently than once every four months, FDA must hold discussions with representatives of veterinary, patient, and consumer advocacy groups to receive their suggestions and discuss their views on the reauthorization. After negotiations with industry are completed, FDA is required to present the recommendations to the specified congressional committees, publish the recommendations in the Federal Register, provide a 30-day public comment period, hold another public meeting to receive views from stakeholders, and revise the recommendations as necessary. Minutes of all negotiation meetings between FDA and industry are required to be posted on the FDA website.54
Proposed AGDUFA III
FDA has published on its website a summary of the AGDUFA III proposed statutory changes and proposed bill language, as well as a notice in the Federal Register announcing the availability of the AGDUFA III draft recommendations.55 The draft AGDUFA III agreement proposes to continue several commitments from AGDUFA II:
- FDA will continue to allow two-phased Chemistry, Manufacturing, and Controls technical section submissions under the JINAD process;
- FDA and industry remain committed to improving the review and business processes to facilitate the scheduling and conducting of preapproval inspections (PAIs); and
- FDA and industry agree to the importance of using both formal meetings and informal communication to ensure high quality submissions so that performance goals can be achieved.
The draft AGDUFA III agreement also proposes several changes to AGDUFA II, specifically to:
- require all submissions to be electronic beginning in FY2019;
- modify the inflation adjuster from a fixed 4% to a variable inflation adjuster calculated using payroll cost and benefits and the Consumer Price Index less food and energy;
- continue using the workload adjustment, as calculated per CVM policy, except to reset the base years to FY2014 through FY2018, with no workload adjustment for FY2019. The proposed FY2019 baseline for AGDUFA III is $18,336,340. For FY2020-FY2023, annual statutory revenue amounts would be further adjusted for inflation and workload;
- eliminate the final year offset provision and making any excess collections available to "enhance the review process in real time;" and
- authorize the Secretary, when setting fees, to reduce a calculated workload adjustment, as specified.
According to the draft agreement, the planned total five-year revenue for AGDUFA III is $95 million, compared to $38.1 million for AGDUFA II and $27.1 million for AGDUFA I.56
- Table 7 shows the proposed AGDUFA III performance goals compared to AGDUFA II.
|
Submission Type |
Performance goal (in days): Act on 90% within: |
|
|
AGDUFA II |
AGDUFA III |
|
|
Original ANADAs and Reactivations |
270 |
240 |
|
Administrative ANADAs |
100 |
60 |
|
Manufacturing Supplemental ANADAs and Reactivations |
270 |
180 |
|
JINAD Studies |
270 |
180 |
|
JINAD Protocols |
100 |
75 |
Source: 82 Federal Register 49377, October 25, 2017. FDA Presentation, "Animal Generic Drug User Fee Act Reauthorization (AGDUFA III) FY2019 – FY2023," Public Meeting November 2017, https://www.fda.gov/downloads/ForIndustry/UserFees/AnimalGenericDrugUserFeeActAGDUFA/UCM585851.pdf.
According to the agency's section by section summary of the proposed statutory changes, the proposed AGDUFA III, in addition to codifying several of the proposals in the draft agreement, would amend the definition of the "process for the review of abbreviated applications for generic new animal drugs" to include processing of Freedom of Information Act requests, and would add an exemption from fees for certain labeling supplements (to add the number of the approved application to the labeling).57
Appendix A. FDA Animal Drug User Fee Programs: Selected Legislative History
|
ADUFA |
AGDUFA |
|||||
|
Original authorizing law |
Animal Drug User Fee Act, 2003 (ADUFA I; P.L. 108-130) |
Animal Generic Drug User Fee Act, 2008 (AGDUFA I; P.L. 110-316) |
||||
|
FFDCA sections |
§§739, 740, and 740A [21 U.S.C. §§379j-11, -12, and -13] |
§§741-742 [21 U.S.C. §§379j-21 and -22] |
||||
|
Reauthorizations |
|
|
||||
|
Percent of program budget paid by user fees in FY2016 |
34% |
59% |
||||
|
Total Full-time Equivalents (FTEs) in FY2016 |
319 |
71 |
||||
|
Fee schedule for FY2018 |
Application (NADA) fee |
|
Abbreviated Application fee except those subject to §512(d)(4) |
|
||
|
Supplemental w/ safety or effectiveness data |
|
Abbreviated Application fee for those subject to §512(d)(4)a |
|
|||
|
Product fee |
|
Product fee |
|
|||
|
Establishment fee |
|
Sponsor fee (100%)b |
|
|||
|
Sponsor fee |
|
Sponsor fee (75%) |
|
|||
|
|
Sponsor fee (50%) |
|
||||
Source: FFDCA §§739-742. 82 FR 35957, August 2, 2017. 82 FR 35966, August 2, 2017. FDA, "FY2016 ADUFA Financial Report Required by the Animal Drug User Fee Act of 2003." FDA, "FY2016 AGDUFA Financial Report Required by the Animal Generic Drug User Fee Act of 2008."
a. FFDCA §512(d)(4) refers to certain combination animal drugs.
b. The sponsor fee is tiered based on the number of approved ANADAs the sponsor of an application holds. A sponsor with more than 6 approved ANADAs would pay 100% of the sponsor fee. A sponsor with more than 1 but less than 7 approved ANADAs would pay 75% of the sponsor fee, and a sponsor with 1 or fewer ANADAs would pay 50% of the fee.
Appendix B. User Fees and Appropriations
Appendix C. Animal Drug Program Activities
|
FY 2009 Actual |
FY 2010 Actual |
FY 2011 Actual |
FY 2012 Actual |
FY 2013 Actual |
FY 2014 Actual |
FY 2015 Actual |
FY 2016 Actual |
FY 2017 Est. |
|
|
New Animal Drug Submissions |
|||||||||
|
New Animal Drug Applications (NADAs) |
|||||||||
|
Received |
12 |
12 |
11 |
14 |
4 |
18 |
12 |
30 |
31 |
|
Completed |
10 |
13 |
13 |
13 |
6 |
14 |
10 |
20 |
20 |
|
Approved |
7 |
11 |
12 |
11 |
6 |
12 |
8 |
16 |
16 |
|
Pending |
6 |
3 |
1 |
2 |
0 |
4 |
3 |
13 |
24 |
|
New Animal Drug Application Supplements |
|||||||||
|
Received |
521 |
552 |
538 |
417 |
409 |
474 |
514 |
544 |
675 |
|
Completed |
545 |
493 |
606 |
458 |
410 |
455 |
516 |
443 |
550 |
|
Approved |
399 |
344 |
497 |
386 |
334 |
390 |
407 |
333 |
400 |
|
Pending |
137 |
212 |
142 |
101 |
99 |
118 |
116 |
217 |
342 |
|
Investigational New Animal Drug (INAD) Files |
|||||||||
|
Received |
2,812 |
3,377 |
2,720 |
2,372 |
3,560 |
2,782 |
3,734 |
3,198 |
3,500 |
|
Completed |
2,758 |
3,088 |
3,050 |
2,388 |
3,538 |
2,853 |
3,805 |
3,156 |
3,250 |
|
Pending |
409 |
702 |
361 |
345 |
402 |
331 |
335 |
627 |
877 |
|
Generic Animal Drug Submissions |
|||||||||
|
Abbreviated New Animal Drug Applications (ANADAs) |
|||||||||
|
Received |
16 |
21 |
23 |
37 |
38 |
31 |
25 |
22 |
50 |
|
Completed |
40 |
32 |
30 |
26 |
46 |
35 |
32 |
23 |
45 |
|
Approved |
9 |
10 |
6 |
8 |
24 |
18 |
20 |
20 |
20 |
|
Pending |
34 |
25 |
18 |
29 |
21 |
17 |
10 |
9 |
14 |
|
Abbreviated New Animal Drug Application Supplements |
|||||||||
|
Received |
144 |
187 |
199 |
177 |
193 |
221 |
227 |
227 |
340 |
|
Completed |
179 |
196 |
238 |
221 |
162 |
229 |
225 |
204 |
300 |
|
Approved |
92 |
112 |
154 |
171 |
127 |
162 |
166 |
123 |
200 |
|
Pending |
170 |
166 |
126 |
82 |
112 |
104 |
116 |
189 |
229 |
|
Generic Investigational New Animal Drug (JINAD) Files |
|||||||||
|
Received |
305 |
271 |
219 |
304 |
498 |
497 |
354 |
502 |
750 |
|
Completed |
327 |
269 |
214 |
305 |
489 |
476 |
358 |
469 |
675 |
|
Pending |
55 |
67 |
57 |
56 |
66 |
87 |
78 |
476 |
551 |
Source: Annual FDA Congressional Budget Justifications, FY2011 through FY2018, Animal Drugs and Feeds sections, Animal Drugs and Feeds Program Activity Data (PAD) tables.
Author Contact Information
Footnotes
| 1. |
CRS Report RL34459, Animal Drug User Fee Programs, by [author name scrubbed]. For information about the human medical products user fees, see CRS Report R44750, FDA Human Medical Product User Fee Programs: In Brief, by [author name scrubbed] et al. |
| 2. |
See comments of Dr. Richard Carnavale, Vice President for Scientific and Regulatory Affairs, Animal Health Institute, at FDA public meeting on ADUFA reauthorization, March 11, 2008, at http://www.fda.gov/cvm/ADUFA032008Transcript.htm. Many of these companies are members of the Animal Health Institute, the trade association that represents their interests, at http://www.ahi.org/. |
| 3. |
FFDCA §740A(d)(6). Food and Drug Administration (FDA), "Animal Drug User Fee Act (ADUFA)," https://www.fda.gov/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/default.htm. |
| 4. |
FFDCA §742(d)(6). FDA, "Animal Generic Drug User Fee Act (AGDUFA)," https://www.fda.gov/ForIndustry/UserFees/AnimalGenericDrugUserFeeActAGDUFA/default.htm. |
| 5. |
For brand-name animal drugs, FFDCA Section 739(8); 21 U.S.C. §379j–11(8). For generic animal drugs, FFDCA Section 741(k)(10); 21 U.S.C. 379j–21(k)(10). |
| 6. |
FDA, "Office of New Animal Drug Evaluation," https://www.fda.gov/AboutFDA/CentersOffices/OfficeofFoods/CVM/WhatWeDo/ucm077923.htm. |
| 7. |
FDA, "Environmental Impact Considerations," https://www.fda.gov/AnimalVeterinary/DevelopmentApprovalProcess/EnvironmentalAssessments/default.htm. |
| 8. |
FDA, "Minor Use/Minor Species," https://www.fda.gov/AnimalVeterinary/DevelopmentApprovalProcess/MinorUseMinorSpecies/default.htm. |
| 9. |
FDA, "Animal Drugs Marketed as Animal Devices," Post-Market Monitoring of Approved Animal Drugs, https://www.fda.gov/AnimalVeterinary/GuidanceComplianceEnforcement/ComplianceEnforcement/UnapprovedAnimalDrugs/ucm229088.htm. |
| 10. |
FDA, "Generic Animal Drug and Patent Term Restoration Act (GADPTRA)," https://www.fda.gov/AnimalVeterinary/GuidanceComplianceEnforcement/ActsRulesRegulations/ucm049100.htm. |
| 11. |
21 C.F.R 25. |
| 12. |
FFDCA §512(b)(1)(G). When an animal-based food is found to contain an unacceptable or "violative" residue, FDA considers the product to be adulterated under the FFDCA. |
| 13. |
For more information see FDA, "The Ins and Outs of Extra-Label Drug Use in Animals: A Resource for Veterinarians," https://www.fda.gov/animalveterinary/resourcesforyou/ucm380135.htm. |
| 14. |
FDA, "Antimicrobial Resistance," https://www.fda.gov/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/default.htm. The term "antimicrobial" is often used synonymously with "antibiotic," the latter being a drug that affects bacteria. Antimicrobial drugs include antibiotics as well as antiviral and antifungal drugs. Each type of pathogen—bacteria, viruses, and fungi—has been shown to develop resistance to drugs used to treat the infections they cause. |
| 15. |
FDA, "Timeline of FDA Action on Antimicrobial Resistance," https://www.fda.gov/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/ucm438426.htm. |
| 16. |
CRS In Focus IF10190, Antibiotic Use in Food Animals: FDA's Current Activities. |
| 17. |
FDA, "FDA Announces Implementation of [Guidance for Industry] #213, Outlines Continuing Efforts to Address Antimicrobial Resistance," press release, January 3, 2017, https://www.fda.gov/AnimalVeterinary/NewsEvents/CVMUpdates/ucm535154.htm. |
| 18. |
FDA, "Veterinary Feed Directive (VFD)," https://www.fda.gov/AnimalVeterinary/DevelopmentApprovalProcess/ucm071807.htm. |
| 19. |
FDA, "Antimicrobial Drug Sales/Distribution Summary Data," https://www.fda.gov/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/default.htm#Sales%20Data. |
| 20. |
FDA, "Questions and Answers: Summary Report on Antimicrobials Sold or Distributed for Use in Food-Producing Animals," https://www.fda.gov/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/ucm236149.htm. |
| 21. |
FY2018 FDA Justification of Estimates for Appropriations Committees, All Purpose Table, https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/BudgetReports/UCM566294.pdf. |
| 22. |
FDA, "About the Center for Veterinary Medicine (CVM)," https://www.fda.gov/AboutFDA/CentersOffices/OfficeofFoods/CVM/default.htm. |
| 23. |
FDA, "FY2016 AGDUFA Financial Report Required by the Animal Generic Drug User Fee Act of 2008," Appendix D, https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/FinancialReports/AGDUFA/UCM550547.pdf. |
| 24. |
The FDA ADUFA financial reports do not provide this breakdown for FY2004. FDA, "FY2014 ADUFA Financial Report Required by the Animal Drug User Fee Act," https://wayback.archive-it.org/7993/20170113204610/http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/FinancialReports/ADUFA/UCM447958.pdf. |
| 25. |
FDA, "FY2016 ADUFA Financial Report Required by the Animal Drug User Fee Act of 2003," https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/FinancialReports/ADUFA/UCM550684.pdf. |
| 26. |
FDA, "FY2016 AGDUFA Financial Report Required by the Animal Generic Drug User Fee Act of 2008," https://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/FinancialReports/AGDUFA/UCM550547.pdf. |
| 27. |
CRS Report RL34459, Animal Drug User Fee Programs. |
| 28. |
Application fees for NADA supplements are equal to 50% of the NADA fee. |
| 29. |
As defined in 21 C.F.R 558.3(b)(3)-(4) or any successor regulation. |
| 30. |
FDA, ADUFA Financial Reports, https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/FinancialReports/ADUFA/default.htm. |
| 31. |
FFDCA §740(f)(1) and FFDCA §740(g)(2)(A)(ii). |
| 32. |
FFDCA §740(g)(2)(A)(i). |
| 33. |
FFDCA §740A. |
| 34. |
FDA, "FY 2016 Performance Report to Congress for the Animal Drug User Fee Act," p. 11, undated, https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/ucm537556.htm. |
| 35. |
FDA, "FY 2016 Performance Report to Congress for the Animal Drug User Fee Act," Executive Summary, undated, https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/ucm537556.htm. |
| 36. |
FDA, "ADUFA Meetings," https://www.fda.gov/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/ucm042891.htm. |
| 37. |
FDA "ADUFA IV: Summary of the Proposed Changes to the [FFDCA] and Proposed Bill Language," https://www.fda.gov/downloads/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/UCM581830.pdf. For the draft recommendations, see "Animal Drug User Fee Act; Recommendations; Request for Comments; Extension of Comment Period," 82 FR 49380, October 25, 2017. |
| 38. |
FDA, "Drug Residues," https://www.fda.gov/AnimalVeterinary/GuidanceComplianceEnforcement/ComplianceEnforcement/ucm264049.htm. |
| 39. |
This refers to application for a MUMS drug. See FDA, "Conditional Approval Explained: A Resource for Veterinarians," https://www.fda.gov/AnimalVeterinary/ResourcesforYou/ucm413948.htm. |
| 40. |
FDA, "ADUFA IV: Summary of the Proposed Changes to the [FFDCA] and Proposed Bill Language," https://www.fda.gov/downloads/ForIndustry/UserFees/AnimalDrugUserFeeActADUFA/UCM581830.pdf. While the Mutual Recognition Agreement is between the US and European Union, FDA conducts an assessment of each country's regulatory authority individually. According to an FDA press release from October 31, 2017, FDA has determined that it would "recognize eight European drug regulatory authorities as capable of conducting inspections of manufacturing facilities that meet FDA requirements," including France and the United Kingdom. See https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm583057.htm. "The scope of the Agreement covers a broad range of human drugs and biologics and veterinary drugs with specific exclusions. Veterinary products are not immediately included within the scope of the agreement, but will be considered for inclusion within the product coverage of the agreement by no later than July 15, 2019," https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofGlobalRegulatoryOperationsandPolicy/UCM544394.pdf. |
| 41. |
ADUFA IV proposes to amend FFDCA §502 (Misbranded drugs and devices) by adding a subsection (w), which would require the labeling of an approved animal drug to include on its labeling the application number. This would not apply to labeling for certain medicated feeds. |
| 42. |
The FDA must, within that time, either issue an order approving the application, or offer the sponsor a notice of opportunity for hearing regarding the agency's finding, pursuant to FFDCA §512(d)(1), of a basis for withholding approval. |
| 43. |
Testimony of Bernadette M. Dunham, Director, FDA CVM, before the House Committee on Energy and Commerce, Subcommittee on Health, hearing on "Committee Prints on Administration Legislative Proposals on the Animal Drug User Fee Act Amendments of 2008 and the Animal Generic Drug User Fee Act of 2008," June 5, 2008, 110th Cong., 2nd Sess., Washington, DC. |
| 44. |
A supplemental abbreviated application refers to a sponsor's request to change the conditions of an approved ANADA (e.g., product manufacturing changes). |
| 45. |
A JINAD submission refers to the submission of information by the sponsor of a generic new animal drug, for the purpose of evaluation of safety or effectiveness by FDA, in the event of the filing of an ANADA or a supplemental application for such drug. |
| 46. |
Application fees for ANADAs for certain combination products subject to the criteria in FFDCA §512(d)(4) and submitted on or after October 1, 2013 are equal to 50% of the ANADA fee. |
| 47. |
The sponsor fee is tiered based on the number of approved ANADAs the sponsor holds. A sponsor with more than 6 approved ANADAs would pay 100% of the sponsor fee. A sponsor with more than 1 but less than 7 approved ANADAs would pay 75% of the sponsor fee, and a sponsor with 1 or fewer ANADAs would pay 50% of the fee. |
| 48. |
FFDCA §741(d). |
| 49. |
FDA, AGDUFA Financial Reports, https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/FinancialReports/AGDUFA/default.htm. |
| 50. |
FFDCA §741(f)(1) and FFDCA §741(g)(2)(A)(ii). |
| 51. |
FFDCA §741(g)(2)(A)(i). |
| 52. |
"FY2013 Performance Report to Congress for the Animal Generic Drug User Fee Act," https://wayback.archive-it.org/7993/20170114024611/http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/UCM384050.pdf. FDA Presentation, "Animal Generic Drug User Fee Act Reauthorization (AGDUFA III) FY2019 – FY2023," Public Meeting November 2017, https://www.fda.gov/downloads/ForIndustry/UserFees/AnimalGenericDrugUserFeeActAGDUFA/UCM585851.pdf. |
| 53. |
Ibid. |
| 54. |
FDA, "AGDUFA Meetings," https://www.fda.gov/ForIndustry/UserFees/AnimalGenericDrugUserFeeActAGDUFA/ucm270232.htm. |
| 55. |
FDA "AGDUFA III: Summary of the Proposed Changes to the [FFDCA] and Proposed Bill Language," https://www.fda.gov/downloads/ForIndustry/UserFees/AnimalGenericDrugUserFeeActAGDUFA/UCM581832.pdf. For the draft recommendations, see "Animal Generic Drug User Fee Act; Recommendations; Request for Comments; Extension of Comment Period," 82 FR 49377, October 25, 2017. |
| 56. |
82 Federal Register 49379, October 25, 2017. |
| 57. |
AGDUFA III proposes to amend FFDCA §502 (Misbranded drugs and devices) by adding a subsection (w), which would require the labeling of an approved animal drug to include on its labeling the application number. |