Prescription Drug User Fee Act (PDUFA): 2017 Reauthorization as PDUFA VI
The Prescription Drug User Fee Act (PDUFA, now called PDUFA I) was reauthorized as PDUFA VI by the Food and Drug Administration Reauthorization Act of 2017 (FDARA, P.L. 115-52). First passed by Congress in 1992, PDUFA gave the Food and Drug Administration (FDA) the authority to collect fees from the pharmaceutical industry and to use the revenue to support “the process for the review of human drug applications.” FDA regulates the safety and effectiveness of drug and biological products sold in the United States. Prior to marketing a drug, a manufacturer must submit to FDA a new drug application (NDA) or a biologics license application (BLA) demonstrating that the product is safe and effective for its intended use.
FDA’s review of NDAs and BLAs is funded through a combination of annual discretionary appropriations from Congress and user fees collected from the pharmaceutical industry. Prior to FDARA, Congress had last reauthorized PDUFA, for a five-year period, through September 30, 2017, via Title I of the Food and Drug Administration Safety and Innovation Act (FDASIA, P.L. 112-144).
User fees provided 65% of the Human Drugs Program funding for FY2016, accounting for 3,351 full-time equivalent employees. Therefore, as each reauthorization deadline approaches, FDA, industry groups, and many Members of Congress generally see PDUFA and the other human medical product user fees as must-pass legislation. Congress originally intended PDUFA to diminish the backlog of new drug applications at FDA and shorten the time from submission to decision. The general view is that PDUFA has succeeded. FDA has added review staff and reduced its review times. Throughout the reauthorizations, stakeholders (e.g., FDA, industry, and patient groups) have raised different concerns in the context of PDUFA, resulting in changes to the scope of activities covered by PDUFA. For example,
PDUFA II expanded the user fee program’s scope to include activities related to the investigational phases of a new drug’s development, and to increase FDA communications with industry and consumer groups.
PDUFA III again expanded the scope of activities that user fees could support to include a three-year postapproval period.
PDUFA IV removed the three-year limitation on postapproval activities and concentrated on new measures concerning postmarket drug safety.
PDUFA V and VI maintained the scope of activities that PDUFA fees could support, and these fees now generally cover the full lifecycle of the drug product. Each reauthorization has connected prescription drug user fees to performance goals and targets, negotiated between FDA and the pharmaceutical industry. Over time, these goals have changed to reflect advancements in the regulatory science surrounding drug development.
Similar to previous reauthorizations, the PDUFA VI package consists of two parts: (1) statutory language that reauthorizes the program and (2) the performance goals and procedures agreement between FDA and industry for FY2018 through FY2022.
Title I of FDARA reauthorizes PDUFA, allowing FDA to collect fees and use the revenue to support specified activities for the review of prescription brand-name drugs and biologics. Additionally, FDARA includes eight other titles that reauthorize the medical device, generic drug, and biosimilar user fee programs through FY2022; modify the drug and device regulatory processes to encourage the development of drugs and devices for pediatric use; amend the law regarding medical device, prescription drug, and generic drug regulation; and make changes in several cross-cutting areas, such as annual reporting on inspection and analysis of use of funds.
Prescription Drug User Fee Act (PDUFA):
2017 Reauthorization as PDUFA VI
Jump to Main Text of Report
Contents
- Introduction
- Origin of Prescription Drug User Fees
- Current Law
- FDA Drug Review and PDUFA Coverage
- Prescription Drug User Fees
- Fee Types
- Fee Adjustments
- Conditions (or Triggers)
- PDUFA Impact on Review Time and FDA Budget
- Review Time
- Budget
- PDUFA VI Package
Figures
Tables
Summary
The Prescription Drug User Fee Act (PDUFA, now called PDUFA I) was reauthorized as PDUFA VI by the Food and Drug Administration Reauthorization Act of 2017 (FDARA, P.L. 115-52). First passed by Congress in 1992, PDUFA gave the Food and Drug Administration (FDA) the authority to collect fees from the pharmaceutical industry and to use the revenue to support "the process for the review of human drug applications." FDA regulates the safety and effectiveness of drug and biological products sold in the United States. Prior to marketing a drug, a manufacturer must submit to FDA a new drug application (NDA) or a biologics license application (BLA) demonstrating that the product is safe and effective for its intended use.
FDA's review of NDAs and BLAs is funded through a combination of annual discretionary appropriations from Congress and user fees collected from the pharmaceutical industry. Prior to FDARA, Congress had last reauthorized PDUFA, for a five-year period, through September 30, 2017, via Title I of the Food and Drug Administration Safety and Innovation Act (FDASIA, P.L. 112-144).
User fees provided 65% of the Human Drugs Program funding for FY2016, accounting for 3,351 full-time equivalent employees. Therefore, as each reauthorization deadline approaches, FDA, industry groups, and many Members of Congress generally see PDUFA and the other human medical product user fees as must-pass legislation. Congress originally intended PDUFA to diminish the backlog of new drug applications at FDA and shorten the time from submission to decision. The general view is that PDUFA has succeeded. FDA has added review staff and reduced its review times. Throughout the reauthorizations, stakeholders (e.g., FDA, industry, and patient groups) have raised different concerns in the context of PDUFA, resulting in changes to the scope of activities covered by PDUFA. For example,
- PDUFA II expanded the user fee program's scope to include activities related to the investigational phases of a new drug's development, and to increase FDA communications with industry and consumer groups.
- PDUFA III again expanded the scope of activities that user fees could support to include a three-year postapproval period.
- PDUFA IV removed the three-year limitation on postapproval activities and concentrated on new measures concerning postmarket drug safety.
PDUFA V and VI maintained the scope of activities that PDUFA fees could support, and these fees now generally cover the full lifecycle of the drug product. Each reauthorization has connected prescription drug user fees to performance goals and targets, negotiated between FDA and the pharmaceutical industry. Over time, these goals have changed to reflect advancements in the regulatory science surrounding drug development.
Similar to previous reauthorizations, the PDUFA VI package consists of two parts: (1) statutory language that reauthorizes the program and (2) the performance goals and procedures agreement between FDA and industry for FY2018 through FY2022.
Title I of FDARA reauthorizes PDUFA, allowing FDA to collect fees and use the revenue to support specified activities for the review of prescription brand-name drugs and biologics. Additionally, FDARA includes eight other titles that reauthorize the medical device, generic drug, and biosimilar user fee programs through FY2022; modify the drug and device regulatory processes to encourage the development of drugs and devices for pediatric use; amend the law regarding medical device, prescription drug, and generic drug regulation; and make changes in several cross-cutting areas, such as annual reporting on inspection and analysis of use of funds.
2017 Reauthorization as PDUFA VI
Introduction
In 1992, the Prescription Drug User Fee Act (PDUFA, now called PDUFA I) gave the Food and Drug Administration (FDA) the authority to collect fees from the pharmaceutical industry and to use the revenue to support "the process for the review of human drug applications."1 That authority, which expired in 1997, has been renewed on five subsequent occasions, by PDUFA II (1997), PDUFA III (2002), PDUFA IV (2007), PDUFA V (2012), and PDUFA VI (2017). The most recent reauthorization was Title I of the Food and Drug Administration Reauthorization Act (FDARA, P.L. 115-52), which extends the user fee program through September 30, 2022.
For FY2017, 41% of FDA's enacted total program level came from user fees;2 however, user fee revenue provided 63% of FDA's Human Drugs Program budget.3 PDUFA revenue also contributed to the Biologics Program4 and agency-wide headquarters and rent budgets.
Congress first passed PDUFA to supplement the FDA budget outside of direct appropriations from Congress. The added funds were intended to enable the agency to increase its staff so it could finish new drug application reviews sooner, allowing both earlier patient access to new drugs and earlier industry earnings on those drugs. PDUFA I amended the Federal Food, Drug, and Cosmetic Act (FFDCA) to establish the authority and the process for collecting and using industry fees;5 it also required FDA and industry representatives to agree on the performance goals and procedures that the PDUFA revenue would support.
This report describes (1) the origin of prescription drug user fees, (2) current law as amended by PDUFA VI, (3) the impact of PDUFA on FDA application review time and the agency's Human Drugs Program budget, and (4) PDUFA VI reauthorization process.
Origin of Prescription Drug User Fees
In the late 1980s, the median time for FDA to approve a new drug application (NDA) was 29 months. Industry, consumer groups, and FDA agreed that the time from submission of a drug or biologics application to FDA's decision was unacceptably long. Patient advocates argued that a drug in review—and therefore not available for sale—could be the difference between life and death. Manufacturers argued that prolonged review times affected their ability to recoup the costs of research and development. During PDUFA I consideration, FDA estimated that each one-month delay in a review's completion cost a manufacturer an average of $10 million.6
FDA argued that it needed more scientists to review incoming drug applications, as well as backlogged applications, and that it had insufficient appropriations to hire additional scientists to conduct these reviews. For decades, FDA had asked Congress for permission to implement user fees. The pharmaceutical industry generally opposed them, believing the funds might go into the Treasury to reduce federal debt rather than help fund drug reviews.7
The pharmaceutical industry's opposition to user fees was mitigated, thus clearing a path for the 1992 law, when then FDA commissioner David Kessler worked out an arrangement that met two industry demands: (1) performance goals, which would set target completion times for various review processes, and (2) the promise that these fees would supplement—rather than replace—funding that Congress appropriated to FDA. Those steps helped persuade industry groups that the fees would reduce review times—and paved the way for Congress to authorize a revenue source that FDA had sought for over 20 years.
Current Law
PDUFA I—and the subsequent PDUFA II, PDUFA III, PDUFA IV, PDUFA V, and PDUFA VI—authorized the collection of prescription drug user fees and the use of that revenue for specified activities.
|
PDUFA and Its Authorizations |
|
PDUFA I (1993-1997) Prescription Drug User Fee Act P.L. 102-571, October 29, 1992 |
|
PDUFA II (1998-2002) Title I of the Food and Drug Administration Modernization Act (FDAMA) P.L. 105-115, November 21, 1997 |
|
PDUFA III (2003-2007) Title V of the Public Health Security and Bioterrorism Preparedness and Response Act of 2002 P.L. 107-188, June 12, 2002 |
|
PDUFA IV (2008-2012) Title I of the FDA Amendments Act of 2007 (FDAAA) P.L. 110-85, September 27, 2007 |
|
PDUFA V (2013-2017) Title I of the FDA Safety and Innovation Act (FDASIA) P.L. 112-144, July 9, 2012 |
|
PDUFA VI (FY2018-FY2022) Title I of the FDA Reauthorization Act (FDARA) P.L. 115-52, August 18, 2017 |
FDA Drug Review and PDUFA Coverage
Prior to marketing a drug, a manufacturer must submit to FDA a new drug application (NDA) demonstrating that the drug is safe and effective for its intended use. FDA reviews each NDA with three major concerns: (1) safety and effectiveness in the drug's proposed use, (2) appropriateness of the proposed labeling, and (3) adequacy of manufacturing methods to ensure the drug's identity, strength, quality, and purity.8
PDUFA I authorized FDA to use the fee revenue to fund the "process for the review of human drug applications" and defined what that process encompassed. With subsequent reauthorizations, Congress has amended that definition to expand the scope of activities covered by PDUFA.9 The upper portion of Figure 1 depicts the research and development path of a new drug, from basic research through preclinical development and testing on animals, clinical development in trials on human subjects as an investigational new drug (IND), FDA review of the NDA, and, finally, the postapproval period in which the drug is marketed.10 The figure's lower portion illustrates the segments of this path during which FDA may use PDUFA revenue to support its activities and how the scope of those activities has been expanded with subsequent reauthorizations.11
|
Figure 1. Drug Research and Development Path and PDUFA Coverage |
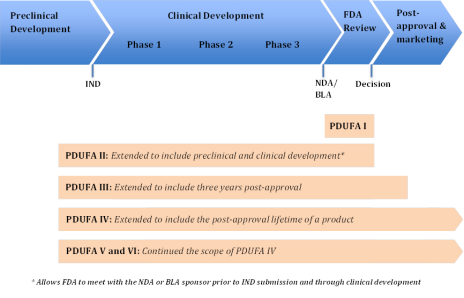 |
|
Source: Prepared by CRS. Notes: BLA = biologics license application, FDA = Food and Drug Administration, IND = investigational new drug, NDA = new drug application, and PDUFA = Prescription Drug User Fee Act. For an explanation of the phases of clinical drug development, see CRS Report R41983, How FDA Approves Drugs and Regulates Their Safety and Effectiveness. |
- PDUFA I (FY1993-FY1997). PDUFA I allowed fee revenue to fund "activities necessary for the review of human drug applications and supplements." In addition to the actual review of applications, it covered activities such as letters from FDA to applicants outlining deficiencies in their applications, facility inspections as part of pending approval applications, and monitoring of research necessary for the review of applications. All those activities fit within the timeframe from when a manufacturer submits an NDA until FDA makes its decision on that application.12
- PDUFA II (FY1998-FY2002). PDUFA II expanded the range of activities for which FDA could use prescription drug user fee revenue to include those related to preclinical and clinical trial phases of a new drug's development.13
- PDUFA III (FY2003-FY2007). PDUFA III extended the range of activities for which FDA could use prescription drug user fee revenue to include three years into the postapproval and marketing period. It allowed FDA to use PDUFA revenue for the collection, development, and review of postmarket safety information for up to three years postapproval (for drugs approved after October 1, 2002). That change allowed the agency to double the number of staff monitoring side effects of drugs already on the market. It also allowed FDA to use fees to develop databases documenting drug use.14
- PDUFA IV (FY2008-FY2012). PDUFA IV removed the three-year limitation on postapproval activities and again expanded the list of postmarket safety activities that the fees could support. New items on the list included developing and using adverse-event data-collection systems, including information technology systems; developing and using improved analytical tools to assess potential safety problems, including access to external databases; implementing and enforcing new FFDCA requirements relating to postapproval studies, clinical trials, labeling changes, and risk evaluation and mitigation strategies; and managing adverse event reports.
- PDUFA V (FY2013-FY2017) and PDUFA VI (FY2018-FY2022). PDUFA V and VI did not change the definition of the "process for the review of human drug applications," thus maintaining the PDUFA IV scope of activities that fees could support.
PDUFA I connected prescription drug user fees to performance goals and targets. FDA negotiated those goals and targets with the pharmaceutical industry and presented them to Congress in the form of a letter from the Department of Health and Human Services Secretary (the Secretary), to which the legislation referred without putting the letter's language directly into law (the FFDCA). PDUFA II and III continued that procedure, again referring to the letter ("PDUFA Reauthorization Performance Goals and Procedures"). However, in 2007, PDUFA IV codified the requirements for a goals letter, consultation and public communication, and other processes as FFDCA Section 736B.15
Prescription Drug User Fees
Fee Types
Each five-year reauthorization sets a total amount of fee revenue for the first year and provides a formula for annual adjustments to that total based on inflation and workload changes. PDUFA I through V had required that three types of fees each contribute one-third of the fee revenue every year: application fees, establishment fees, and product fees.16 PDUFA VI establishes a new user fee structure, eliminating the product and establishment fees and adding a program fee, to provide 80% of the total fee revenue.17 PDUFA VI continues the application fee, to provide 20% of the total fee revenue, while eliminating the fee for a supplemental application.18
- Application fee: The sponsor of an application (usually the manufacturer) must pay a fee for FDA review each time it submits an NDA or BLA.
- Program fee: The sponsor of an application must pay an annual program fee for each prescription drug product that is identified in an application.
According to CDER Director Janet Woodcock's March 2017 testimony at a House Energy and Commerce Committee hearing on the then proposed PDUFA VI,
FDA proposes to enhance the program fee structure and related mechanisms, to achieve increased predictability, stability, and efficiency. The current overall PDUFA fee structure and the fee setting process were established in 1992. Both FDA and industry recognize that updating some elements of the fee structure and the fee setting process will enhance administrative efficiency and the predictability and stability of fee amounts and revenues and improve FDA's ability to engage in long-term financial planning...19
Fee Adjustments
Under PDUFA I through V, fee revenues were adjusted for inflation and to reflect changes in FDA's workload for the process for the review of human drug applications.20 The workload adjustment calculation was based on a weighted average of the change in the total number of human drug applications, commercial IND applications, efficacy supplements, and manufacturing supplements submitted. For additional information about fee adjustments prior to PDUFA VI, see CRS Report R42366, Prescription Drug User Fee Act (PDUFA): 2012 Reauthorization as PDUFA V.
PDUFA VI modifies the inflation adjustment calculation, which is a weighted average of the Consumer Price Index (CPI) figure and FDA personnel cost figures.21 Because the scientists, statisticians, and clinicians who review human drug applications at FDA do not reflect average personnel costs and benefits for the region, the inflation adjustment includes two calculations. Costs of FDA personnel compensation and benefits are calculated based on FDA's historical costs for those items. Other FDA costs are calculated based on the local CPI.22
PDUFA VI also replaces the workload adjustment with a capacity planning adjuster intended to better align fees with the workload and existing staff capacity at FDA. More specifically, PDUFA VI requires the Secretary to "obtain, through a contract with an independent accounting or consulting firm, a report evaluating options and recommendations for a new methodology to accurately assess changes in the resource and capacity needs of the process for the review of human drug applications."23 After review of the report and any public comments, the Secretary is required to establish a capacity planning methodology, incorporating approaches and attributes the Secretary finds appropriate, to be effective beginning in the first fiscal year for which fees are set after the methodology is established. In the interim before such capacity planning methodology is effective, the workload adjustment would be based on the product of the annual base revenue for the year, adjusted for inflation, and the adjustment percentage for a fiscal year, as specified.24
PDUFA VI eliminates the final-year adjustment provisions and instead establishes an annual operating reserve adjustment.25 Under this provision, the Secretary may increase the fee revenue and fees "to provide for not more than 14 weeks of operating reserves of carryover user fees for the process for the review of human drug applications," a provision similar to PDUFA V's reference to a three-month reserve. PDUFA VI adds the inverse, requiring that if the Secretary has carryover balances in excess of 14 weeks of operating reserves, then the Secretary must "decrease such fee revenue and fees to provide for not more than 14 weeks of such operating reserves."26
PDUFA VI also establishes an additional direct cost adjustment, requiring the Secretary to further increase the fee revenue and fees by $8.7 million for FY2018. For FY2019 through FY2022, the amount of the additional direct adjustment would be equal to $8.7 million multiplied by the CPI, as specified.27
The total revenue under PDUFA VI for each of fiscal years FY2018 though FY2022 is to equal the sum of
- the annual base revenue ($878.6 million) for the fiscal year;
- the dollar amount equal to the inflation adjustment for the fiscal year;
- the dollar amount equal to the capacity planning adjustment for the fiscal year;
- the dollar amount equal to the operating reserve adjustment for the fiscal year, if applicable;
- the dollar amount equal to the additional direct cost adjustment for the fiscal year; and
- the additional dollar amounts specified for each fiscal year.28
Conditions (or Triggers)
A key element of PDUFA, carried through all reauthorizations, is that user fees are to supplement congressional appropriations, not replace them. The law has included three limiting conditions, known as "triggers," to enforce that goal. FDA may collect and use fees only if (1) FDA's overall Salaries and Expenses direct appropriation equals or exceeds the agency's 1997 Salaries and Expenses appropriation, adjusted for inflation; (2) the fee amounts are provided in the appropriations acts; and (3) the agency spends at least as much from appropriated funds for the review of human drug applications as it spent in FY1997, adjusted for inflation. PDUFA VI does not change these conditions.
PDUFA Impact on Review Time and FDA Budget
Review Time
The approval times for NDAs and BLAs provide a measure of PDUFA's effectiveness in meeting its primary goal: reducing the time between a manufacturer's submission of an NDA/BLA and FDA's approval decision. Under PDUFA I, FDA agreed to specific goals for improving the drug review time and created a two-tiered system of review times: Standard Review and Priority Review. While the goal for standard review is 10 months, a priority review designation means FDA's goal is to take action on an application within 6 months.29 An application may receive priority review designation if it is for a drug that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. Figure 2 shows the median approval times (standard and priority) of new molecular and biologic entities.
According to a December 2016 FDA presentation, as of September 30, 2016, agency data indicated that FDA had met or exceeded 10 of the 12 specified performance goals for applications submitted in FY2015 and was, at the time in FY2016, meeting or exceeding all (12 out of 12) performance goals for FY2016 submissions.30
|
Figure 2. CDER New Molecular Entity (NME) NDA/BLA Median Time to Approval (by standard, priority, and overall review) |
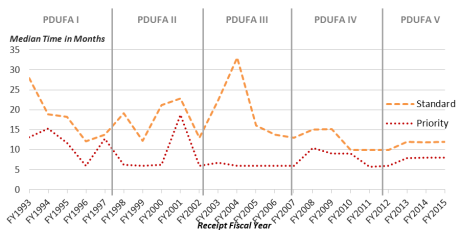 |
|
Source: Figure created by CRS using data from the FDA Presentation "CDER New Drug Review: 2016 Update," December 14, 2016, slide 21, https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM533192.pdf. Notes: Data are current as of December 9, 2016. No FY2016 standard applications had been acted upon at that date. On December 5, 2017, FDA issued a 2017 CDER update; however, this presentation does not include data on approval times, see https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM587690.pdf. |
Budget
Table 1 shows, for the Human Drugs total program level, the relative contributions of the two funding sources—budget authority and user fees—over time. In the first year of PDUFA contributions to the FDA budget, user fee revenue accounted for 9.7% of the Human Drugs total program level. By FY2017, user fee revenue increased to 63% of the Human Drugs program; this includes revenue from PDUFA, generic drug user fees, biosimilar user fees, and outsourcing facility fees.31
Table 1. FDA Human Drugs Program, Fees as a Percentage of Total Program Level for Selected Fiscal Years
(unadjusted dollars)
|
Fiscal Year |
Budget Authority |
Fees |
Total Program Level |
|
|
$ in millions |
$ in millions |
Fees as % of total program level |
$ in millions |
|
|
1994 |
$214.9 |
$23.1 |
9.7% |
$238.0 |
|
1998 |
$199.6 |
$63.1 |
24.0% |
$262.6 |
|
2003 |
$274.1 |
$129.8 |
32.1% |
$403.8 |
|
2008 |
$353.9 |
$327.0 |
48.0% |
$680.9 |
|
2013 |
$438.6 |
$602.1 |
57.8% |
$1,040.6 |
|
2017 |
$492.2 |
$837.1 |
63.0% |
$1,329.3 |
Source: The FY1994 through FY2013 are from the FDA Justification of Estimates for Appropriations Committees documents, FY1996 through FY2017. The FY2017 amounts are from the 2017 Consolidated Appropriations Act (P.L. 115-31).
Notes: FY1994 was the first year that PDUFA revenue was reflected in the FDA budget. FY1998, FY2003, FY2008, and FY2013 were the first fiscal years of PDUFA II, III, IV, and V. Until FY2013, PDUFA fees were the only user fees going to the Human Drugs Program. FY2017 is the most recent year for which data are available.
PDUFA VI Package
The PDUFA VI package consists of two parts: (1) the performance goals and procedures agreement between FDA and industry, which this report refers to as the Commitment Letter or the Agreement (summarized in Appendix A), and (2) statutory language that reauthorizes the program (summarized in Appendix B). The timeline for the development of the Agreement and the reauthorization of the program is shown in Figure 3.
The PDUFA VI reauthorization process began with a public meeting held in July 2015, followed by a 30-day comment period. From September 2015 through February 2016, FDA held meetings with industry and patient and consumer advocacy groups; minutes of the meetings are available on the FDA website.32 On July 15, 2016, FDA published a notice in the Federal Register announcing the availability of the proposed PDUFA VI Commitment Letter, as well as a public meeting to discuss the proposed recommendations for the PDUFA VI reauthorization.33 On April 25, 2017, user fee legislation was introduced in the Senate (S. 934) that would have reauthorized PDUFA and the three other human medical product user fee programs, among other things. On May 11, 2017, the Senate Health, Education, Labor, and Pensions (HELP) Committee ordered the bill to be reported with an amendment in the nature of a substitute. In the House, a version of the FDA Reauthorization Act (H.R. 2430) was introduced on May 16, 2017, and referred to the House Energy and Commerce Subcommittee on Health, which approved it with amendments by voice vote on May 18, 2017. The full committee approved the bill on June 7, 2017. The House passed H.R. 2430 on July 12, 2017, and the Senate passed it on August 3, 2017.
H.R. 2430 was signed into law as FDARA (P.L. 115-52) on August 18, 2017. Title I reauthorizes PDUFA, allowing FDA to collect fees and use the revenue to support specified activities for the review of prescription brand-name drugs. FDARA includes eight other titles. A separate CRS report describes provisions in those titles, which cover reauthorization of the medical device, generic drug, and biosimilar biological product user fees, as well as provisions concerning pediatric drugs and devices, reauthorizations and improvements related to drugs, inspections and regulatory improvements related to devices, improvements to generic drug access, and a set of miscellaneous provisions.34
Appendix A.
Summary of PDUFA VI Agreement:
Performance Goals and Procedures FY2018-FY2022
|
PDUFA VI Commitments |
|
|
I. Ensuring the Effectiveness of the Human Drug Review Program |
|
|
A. Review Performance Goals |
For major types of applications and efficacy and manufacturing supplements, states (1) the time in which FDA agrees, as a goal, to review and act on an application, and (2) the percentage of applications for which FDA agrees to meet that goal. See Table A-2. The Agreement also addresses extensions for certain amendments to applications, efficacy supplements, or additions to the list of facilities that FDA must inspect as part of an application. |
|
B. Program for Enhanced Review Transparency and Communication for NME NDAs and Original BLAs |
FDA will apply "the Program" to the review of all NME NDAs and original BLAs, including resubmissions following a refuse-to-file decision. The Program includes certain parameters: a presubmission meeting (generally not less than two months before planned submission of the application); the expectation that original application submissions are complete; a Day 74 letter sent by FDA to the applicant with a planned date for the internal midcycle review meeting and preliminary plans on whether to hold an advisory committee meeting; review performance goals; midcycle communication; late-cycle communication and advisory committee meetings; and inspections. (FDA's goal is to complete GCP, GLP, and GMP inspections for applications in the Program within 6 months of the receipt date for priority review and 10 months for standard review.) The FDA review team and applicant may also decide on an alternative approach regarding the timing and nature of interactions and information exchange (i.e., a "Formal Communication Plan"). For applications that FDA identifies as meeting an important public health need, the review team "intends to make every effort to conduct an expedited review and act early on the application." Expedited reviews include frequent contact between the applicant and the FDA team through the review process. |
|
C. First Cycle Review Management |
To ensure an efficient and effective first cycle review process, FDA will update its 2005 Good Review Management Principles (GRMP) guidance to include review activities (e.g., the NME Program, REMS) that have been added to the human drug review program since the guidance as finalized; the agency will publish revised draft guidance for public comment by the end of FY2018. |
|
D. Review of Proprietary Names to Reduce Medication Errors |
"To reduce medication errors related to look-alike and sound-alike proprietary names and factors such as unclear label abbreviations, acronyms, dose designations, and error prone label packaging design," FDA will review proprietary names submitted during drug development and along with the NDA/BLA according to timeframes detailed in the Agreement. |
|
E. Major Dispute Resolution |
For disputes over procedural or scientific matters that cannot be resolved at "signatory authority level," the Agreement allows for written appeals to the next two levels, according to specified criteria. FDA agrees to respond to 90% of such appeals within 30 days of their receipt. |
|
F. Clinical Holds |
After a sponsor submits a complete response to a clinical hold, FDA agrees to respond to 90% of such responses within 30 days of receipt. |
|
G. Special Protocol Question Assessment and Agreement |
The Agreement lays out procedures, including timing and criteria for sponsor submissions of a limited number of specific questions about protocol design and regulatory and scientific requirements. "The fundamental agreement here is that having agreed to the design, execution, and analyses proposed in protocols reviewed under this process, the Agency will not later alter its perspective on the issues of design, execution, or analyses unless public health concerns unrecognized at the time of protocol assessment under this process are evident." FDA agrees to complete and return 90% of these special protocol assessments and agreement requests within timeframes, and to track and report the number of such assessments and resubmissions per original special protocol assessment. |
|
H. Meeting Management Goals |
Regarding requests for specific types of meetings (Types A, B, B(EOP), and C, which are described in the Commitment Letter), the Agreement outlines timeframes and notification requirements for requests and meeting schedules, content, attendees, documentation, and minutes. The PDUFA VI Agreement adds criteria for when FDA or the sponsor could provide a written response to questions rather than hold a meeting. FDA agrees to respond to 90% of meeting requests within the specified timeframes. The Agreement adds the requirement that "FDA will publish revised draft guidance on formal meetings between FDA and sponsors no later than September 30, 2018." |
|
I. Enhancing Regulatory Science and Expediting Drug Development |
(1) Promoting Innovation through Enhanced Communication between FDA and Sponsors during Drug Development. FDA will maintain "dedicated drug development communication and training staffs in CDER and CBER." The function of the staff is (1) to serve as liaison that will facilitate interactions between sponsors and each Center, and (2) to provide ongoing training to the review organizations on best practices in communication with sponsors. To enhance timely interactive communication with sponsors during drug development in PDUFA VI, FDA will contract with an independent third party to assess current FDA and sponsor communication practices; convene a public workshop by the end of 2021; consider the third party's recommendations and public feedback, and if FDA determines it to be appropriate, update the current draft or final guidance on "Best Practices for Communication between IND Sponsors and FDA During Drug Development." (2) Ensuring Sustained Success of Breakthrough Therapy Program. FDA and the industry are committed to ensuring the expedited development and review of breakthrough therapies. Additional resources will allow the agency to continue to work closely with sponsors throughout the designation, development, and review processes. (3) Early Consultation on the Use of New Surrogate Endpoints. FDA agrees to consider an early consultation meeting on the feasibility of using a new surrogate endpoint as the primary basis for product approval as a Type C meeting. The PDUFA VI agreement specifies that to qualify for such a consultation, Type C meeting requests must include the complete background package as specified. (4) Advancing Development of Drugs for Rare Diseases. FDA agrees to continue "to advance and facilitate the development and timely approval of drugs and biologics for rare diseases." The RDP staff in CDER will be integrated into review teams for rare disease development programs and application review, and will provide training to CDER and CBER review staff. RDP staff will continue to engage in outreach to stakeholders to provide training on FDA's RDP, will continue to foster collaborations in tool and data development, and will facilitate interactions between stakeholders and FDA review divisions. FDA will include updates on the activities and success of the RDP in the PDUFA annual performance reports and will continue to include information on rare disease approvals in its annual reports on innovative drug approvals. (5) Advancing Development of Drug-Device and Biologic-Device Combination Products Regulated by CBER and CDER. FDA agrees to develop staff capacity across the medical product centers and the Office of Combination Products (OCP) to review and respond to submissions that include combination products. Within the specified timeframes, FDA will "streamline the process for combination product review and improve the Agency's ability to assess workload and allocate resources to the review of combination products." FDA will develop Manuals of Policies and Procedures (MAPPs) and Standard Operating Policy and Procedures (SOPPs) addressing combination product development and review. By specified timeframes, FDA agrees to make available on its website, and update periodically, key points of contact in OCP and the medical product centers to combination product review; establish submission procedures for Human Factors protocols and establish timelines to review and provide comment on the protocols for such Human Factors studies; begin staff training related to development, review, and approval of drug-device and biologic-device combination products reviewed in CBER and CDER; contract with an independent third party to assess current practices for combination product review; and publish draft or update guidance describing considerations related to drug-device and biologic-device combination products, as specified. (6) Enhancing Use of Real World Evidence (RWE) for Use in Regulatory Decision-Making. By the specified time frames, FDA agrees to complete at least one public workshop with key stakeholders (e.g., patients, industry, academia) to gather input into issues related to use of RWE in regulatory decisionmaking; initiate (or fund by contract) "appropriate activities aimed at addressing key outstanding concerns and considerations in the use of RWE for regulatory decision-making"; and issue draft guidance on how RWE can contribute to safety and effectiveness assessments in regulatory submissions. |
|
J. Enhancing Regulatory Decision Tools to Support Drug Development and Review |
(1) Enhancing the Incorporation of the Patient's Voice in Drug Development and Decision-Making. To facilitate development and use of patient- and caregiver-focused methods to inform drug development and decisionmaking, FDA agrees to strengthen staff capacity; develop a series of guidance documents by the specified timelines; create and maintain a repository of publicly available tools on the agency's website; revise existing MAPPs and SOPPs; and conduct a public workshop, through a third party, to gather patient and caregiver experience data. (2) Enhancing Benefit-Risk Assessment in Regulatory Decision-Making. To "further the agency's implementation of structured benefit-risk assessment, including the incorporation of the patient's voice in drug development and decision-making," the agency agrees to, by the specified timeframes, publish an update to the "Structured Approach to Benefit-Risk Assessment in Drug Regulatory Decision-Making" implementation plan, including a report on the progress made in PDUFA V; convene or participate in at least one meeting conducted by a third party to gather stakeholder on key topics; publish draft guidance on benefit-risk assessments for new drugs and biologics; conduct an evaluation of the implementation of the benefit-risk framework in the human drug program; and revise relevant MAPPs and SOPPs to include new approaches that incorporate FDA's benefit-risk framework into the human drug review program. (3) Advancing Model-Informed Drug Development. "To facilitate the development and application of exposure-based, biological, and statistical models derived from preclinical and clinical data sources, herein referred to as 'model-informed drug development' (MIDD) approaches," FDA agrees to develop its expertise and capacity in MIDD approach; convene a series of workshops to identify best practices for MIDD on specified topics; conduct a pilot program for MIDD approaches; publish draft guidance or revise existing guidance on MIDD; and develop or revise relevant MAPPs and SOPPs to incorporate guidelines for the evaluation of MIDD approaches. (4) Enhancing Capacity to Review Complex Innovative Designs. FDA agrees to develop the staff capacity to facilitate appropriate use of complex adaptive, Bayesian, and other novel clinical trial designs. FDA will also conduct a pilot program for "highly adaptive trial designs for which analytically derived properties (e.g., Type 1 error) may not be feasible, and simulations are necessary to determine trial operating characteristics." The agency will announce the pilot program in the Federal Register, select up to two proposals quarterly each year, and convene an internal review group to review select proposals. FDA may use the trials designs developed in the pilot program as case studies, and the agency and sponsor will agree upon what information FDA may share publicly in these case studies. FDA may periodically review and determine whether to adjust aspects of the program. FDA also agrees to convene a public workshop to discuss complex adaptive trial designs; publish draft guidance on complex adaptive trial designs; and revise relevant MAPPs and SOPPs as appropriate. (5) Enhancing Capacity to Support Analysis Data Standards for Product Development and Review. FDA agrees to develop the staff capacity to review and provide feedback to sponsors on the readiness of submitted analysis data sets for statistical review; improve staff capacity to assist with FDA development and updating of therapeutic area user guides (TAUGs); convene a public workshop to advance analysis data standards; collaborate with stakeholders and participate in public workshops on development of data standards, processes, documentation, and continuous improvement of clinical trials and regulatory science; and develop or revise relevant guidance, MAPPs, SOPPs and training associated with standardized analysis datasets and programs used in review. (6) Enhancing Drug Development Tools Qualification Pathway for Biomarkers. FDA agrees to enhance staff capacity to enhance biomarker qualification review; convene a public meeting to discuss biomarker taxonomy; publish draft guidance on proposed taxonomy of biomarker usage and general evidentiary standards for biomarker qualification; develop or revise relevant MAPPs and SOPPs on the biomarker qualification process; and make publicly available on the internet a list of biomarker qualification submissions that are in the qualification process. |
|
K. Enhancement and Modernization of the FDA Drug Safety System |
FDA agrees to continue to use user fees to enhance and modernize the drug safety system. FDA will use user fees to support advancing postmarket drug safety evaluation through enhancement of the Sentinel System and integration into FDA pharmacovigilance activities. Specifically, within the specified timeframes, FDA will enhance its communication with sponsors and the public regarding methodologies for Sentinel queries; evaluate additional ways to facilitate access to Sentinel's distributed data network to conduct safety surveillance; hold a public meeting seeking stakeholder feedback on Sentinel and its system of Active Risk Identification and Analysis (ARIA); establish MAPPs and SOPPs about the planned use of Sentinel; facilitate integration of Sentinel into the human drug review program through staff development and by updating existing SOPPs and MAPPs; develop a comprehensive training program for review staff to ensure they have a working knowledge of Sentinel; and analyze and report on the impact of Sentinel expansion and integration. FDA will use user fees to continue to support the review, oversight, tracking, and communication of postmarket drug safety issues. Specifically, the agency will make improvements to its current processes that capture and track information; update existing MAPPs and SOPPs concerning tracking postmarket safety signals; conduct, or fund by contract, as assessment of how its data systems and processes support review, oversight, and communication of postmarket drug safety issues. |
|
II. Enhancing Management of User Fee Resources |
|
|
A. Resource Capacity Planning and Modernized Time Reporting |
FDA will publish a PDUFA program resource capacity planning and modernized time reporting implementation plan no later than the second quarter of FY2018; staff a resource capacity planning team; and obtain through an independent firm an evaluation report of options and recommendations for a new methodology to accurately assess changes in the resources and capacity needs of the human drug review program. The evaluation report will be published for public comment and upon review of the report and comments, FDA agrees to "implement robust methodologies for assessing resource needs of the program." The agency will also document in the annual PDUFA financial report "how the workload adjuster and resource capacity adjustment fees are being utilized." |
|
B. Financial Transparency and Efficiency |
FDA agrees to contract with an independent third party to conduct an evaluation of PDUFA program resource management during FY2018 to ensure resources are being appropriately administered, allocated, and reported in an efficient and transparent manner, as specified. FDA will publish, within specified timeframes, a PDUFA five-year financial plan and updates, and will convene a public meeting to discuss the five-year financial plan, among other things. |
|
III. Improving FDA Hiring and Retention of Review Staff |
|
|
Completion of Modernization of the Hiring System Infrastructure and Augmentation of System Capacity |
FDA will complete development and implementation of the FTE-based position management system, and will finalize the establishment of an online Position Description (PD) library. For key scientific and technical disciplines, FDA will complete transition to expanded use of a common vacancy announcement and certificate of eligible job applicants that can be used by multiple offices. |
|
Augmentation of Hiring Staff Capacity and Capability |
"FDA will engage a qualified contractor to provide continuous support throughout PDUFA VI to augment the existing FDA HR staff capacity and capabilities." |
|
Complete Establishment of a Dedicated Function to Ensure Needed Scientific Staffing for Human Drug Review Program |
"FDA will complete the establishment of a new dedicated unit within the Office of Medical Products and Tobacco charged with the continuous recruiting, staffing, and retention of scientific, technical and professional staff for the process for the review of human drug applications." The unit will develop and implement scientific staff hiring strategies and plans, and will conduct analyses of compensation and other factors affecting retention of key staff in targeted disciplines. |
|
Set Clear Goals for Human Drug Review Program Hiring |
FDA will establish goals for hiring within the human drug review program staff for the years of PDUFA VI, as specified in table 6 of the agreement. The agency will report on progress toward those goals for FY2018-2022 on a quarterly basis on the FDA website. |
|
Comprehensive and Continuous Assessment of Hiring and Retention |
FDA agrees to contract with a qualified, third-party contractor who will conduct a comprehensive review of agency hiring processes and hiring staff capacity. The agreement specifies timeframes for completion of the initial, interim, final assessments, and public comments. |
|
IV. Information Technology Goals |
|
|
Objective |
FDA "is committed to achieve the long-term goal of improving the predictability and consistency of the electronic submission process (Section IV.B), and enhancing transparency and accountability of FDA information technology related activities (Section IV.C) ... through IT investments that support the PDUFA program" |
|
Improve the Predictability and Consistency of PDUFA Electronic Submission Processes |
FDA agrees to publish and maintain up-to-date documentation for the electronic submission process, as specified. The agency will publish targets for and measure Electronic Submission Gateway (ESG) availability. |
|
Enhance Transparency and Accountability of FDA Electronic Submission and Data Standards Activities |
FDA and industry agree to jointly plan and hold quarterly meetings and to share performance updates prior to each meeting. By the specified timeframes, FDA agrees to hold annual public meetings to seek stakeholder input on issues related to electronic submission; post on the FDA website, at least annually, metric on ESG performance, as specified; and incorporate strategic initiatives in support of PDUFA goals into the FDA IT Strategic Plan. FDA also will collaborate with Standards Development Organizations and stakeholders; publish (and update quarterly) a data standards action plan; and publish and maintain a current FDA Standards Data Catalog. |
|
V. Improving FDA Performance Management |
FDA agrees to conduct the studies described throughout the agreement. |
|
VI. Progress Reporting for PDUFA VI and Continuing PDUFA V Initiatives |
FDA will include in the annual PDUFA Performance Report information about the agency's progress in meeting the commitments specified in sections I.I-K (enhancing regulatory science and expediting drug development, enhancing regulatory decision tools to support drug development and review, and enhancement and modernization of the FDA Drug Safety System), as well as progress in the hiring of new staff used to support the new initiatives in Section III. |
|
VII. Definitions and Explanation of Terms |
See PDUFA VI Agreement for definitions. |
Source: CRS summary of "PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2018 through 2022," https://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM511438.pdf.
|
Application/Supplement |
Standard |
Priority |
|
NME NDAs and original BLAs |
90% in 10 months of the |
90% in 6 months of the |
|
Non NME NDAs |
90% in 10 months of the |
90% in 6 months of the |
|
Class 1 Resubmissions |
90% in 2 months of the |
90% in 2 months of the |
|
Class 2 Resubmission |
90% in 6 months of the |
90% in 6 months of the |
|
Original Efficacy Supplements |
90% in 10 months of the |
90% in 6 months of the |
|
Class 1 Resubmitted Efficacy Supplements |
90% in 2 months of the |
90% in 2 months of the |
|
Class 2 Resubmitted Efficacy Supplements |
90% in 6 months of the |
90% in 6 months of the |
|
Prior Approval |
All Other |
|
|
Original Manufacturing Supplements |
90% in 4 months of the receipt date |
90% in 6 months of the receipt date |
Source: Tables 1 and 2 from "PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2018 through 2022," https://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM511438.pdf.
Appendix B. Provisions in FFDCA Sections 735, 736, and 736B
Table B-1. FFDCA Sections 735, 736, and 736B Relating to Prescription Drug User Fees, as amended by PDUFA VI
|
Summary of Current Law (Reflecting PDUFA VI) |
|
|
FFDCA Sec. 735 [21 U.S.C. 379g]. Definitions. |
|
|
Definitions |
This section defines how several terms are used in this part: human drug application, supplement, prescription drug product, final dosage form, prescription drug establishment, process for the review of human drug applications, costs of resources allocated for the process for the review of human drug applications, adjustment factor, person, active, and affiliate. In particular, it defines the term "process for the review of human drug applications" as activities necessary for the review of human drug applications and supplements; the issuance of action letters; inspection of prescription drug establishments and other facilities; activities necessary for the review of applications for licensure of biological product establishments and for the release of lots of biologics; monitoring of research conducted in connection with the review of human drug applications; and postmarket safety activities, including adverse event data collection systems and development of analytical tools, and enforcement of study and label-change requirements. The term "costs of resources allocated for the process for the review of human drug applications" is defined as "expenses in connection with the process for the review of human drug applications for" FDA officers, employees, contractors, and advisory committees; information management and computer resources; facilities, furniture, equipment, materials and supplies; and collecting user fees and accounting for resources. [Sec. 736(f)(3) adds a limitation: Beginning October 1, 2023, the costs of resources shall include only expenditures for leasing and necessary scientific equipment.] |
|
FFDCA Sec. 736 [21 U.S.C. 379h]. Authority to Assess and Use Drug Fees. |
|
|
(a) Types of fees |
There are two types of fees—application and program—and certain exceptions. |
|
(a)(1) Human drug application fee |
A human drug application fee is assessed for an application for which clinical data with respect to safety or effectiveness are required for approval. The fee for an application that does not require clinical data is half the full application fee. The fee is due at the time of application submission. Exceptions are made for a previously filed application under certain conditions and for a designated orphan drug (unless the application includes an indication for other than a rare disease or condition). |
|
(a)(2) Prescription drug program fee |
A prescription drug program fee is to be paid annually by each person who is named as an applicant in a human drug application, for each prescription drug product named in such application (except for a product whose manufacturer has had no pending application since September 1, 1992). Exceptions are made for certain prescription drug products. Includes a limitation that an applicant shall not be assessed more than five prescription drug program fees for a fiscal year with respect to an approved application. |
|
(b)(1) Fee Revenue Amounts |
The total user fee revenue for each fiscal year is to equal the sum of the annual base revenue for the fiscal year; the dollar amount equal to the inflation adjustment for the fiscal year; the dollar amount equal to the capacity planning adjustment for the fiscal year; the dollar amount equal to the operating reserve adjustment for the fiscal year, if applicable; the dollar amount equal to the additional direct cost adjustment for the fiscal year; and the additional dollar amounts specified for each fiscal year.a |
|
(b)(2) Types of fees |
Of the total prescription drug user fee revenue to be collected each year, 20% is to come from the application fee and 80% is to come from program fees. |
|
(b)(3) Annual base Revenue |
The annual base revenue amount for FY2018 is $878,590,000, to be adjusted annually for FY2019-FY2022. |
|
(c) Adjustments; annual fee setting |
PDUFA VI changed the formula for the workload and inflation adjustment factors. |
|
(c)(1) Inflation adjustment |
The inflation adjustment for a fiscal year is equal to the product of the annual base revenue for a fiscal year (under FFDCA §736(b)(1)(A)) and the inflation adjustment percentage. The inflation adjustment percentage for a fiscal year is to equal the sum of: (1) the average annual percent change in FDA personnel costs (i.e., compensation and benefits) for the first three of the preceding four fiscal years, multiplied by the proportion of personnel compensation and benefits costs to total costs of the process for the review of human drug applications (as defined in FFDCA §735(6)) for the first three years of the preceding four years, and (2) the CPI figure, which is the average annual percent change in the CPI (for DC-MD-VA-WV, all items) for the first three years of the preceding four years of available data multiplied by the proportion of all costs other than personnel compensation and benefits costs to total costs of the process for the review of human drug applications (as defined in FFDCA §735(6)) for the first three years of the preceding four fiscal years. |
|
(c)(2) Capacity planning adjustment |
After the annual base revenue is adjusted for inflation, it must be further adjusted to "reflect changes in the resource capacity needs" for the review of human drug applications. The HHS Secretary is required to establish a capacity planning methodology, after obtaining a report evaluating options and recommendations for a new methodology to assess changes in the resource and capacity needs of the process for the review of human drug applications. The report is to be done under contract with an independent accounting or consulting firm and to be published for public comment. In the interim before such capacity planning methodology is effective, the adjustment shall be based on the product of the annual base revenue for the year (adjusted for inflation) and the adjustment percentage for a fiscal year. The adjustment percentage clause for a fiscal year is the weighted change in the three-year average ending in the most recent year for which data are available, over the three-year average ending in the previous year, for (1) the total number of human drug applications, efficacy supplements, and manufacturing supplements submitted; (2) the total number of active commercial investigational new drug applications; and (3) the total number of formal meetings scheduled by the Secretary, and written responses issued by the Secretary in lieu of such formal meetings, as identified in the Commitment Letter. Such adjustment shall not result in fee revenue for a fiscal year that is less than the sum of the annual-base and inflation-adjustment amounts. The Secretary must publish in the Federal Register the revenue and fees resulting from this adjustment along with the calculation methodologies. |
|
(c)(3) Operating Reserve Adjustment |
The Secretary may increase fee revenue and fees if necessary to provide for not more than 14 weeks of operating reserves of carryover fees for human drug application review. If there are carryover balances in excess of 14 weeks of operating reserves, the Secretary must decrease fee revenue and fees to provide for not more than 14 weeks of such operating reserves. If such an adjustment is made, the rationale must be published in the annual Federal Register notice establishing fee revenue and fees for the fiscal year involved. |
|
(c)(4) Additional Direct Cost Adjustment |
The Secretary must further increase fee revenue and fees by $8,730,000 for FY2018. For FY2019 through FY2022, the amount of the additional direct adjustment is to equal $8,730,000 multiplied by the CPI (for DC-MD-VA-WV, all items) for the most recent year of available data, divided by the CPI for 2016. |
|
(c)(5) Annual fee setting |
The Secretary must establish fees each year, 60 days before the start of each fiscal year, and publish the fee revenue and fees in the Federal Register. |
|
(c)(6) Limit |
For each fiscal year, the total amount of fees, as adjusted, may not exceed the total costs for the resources allocated for the human drug application review process. |
|
(d) Fee Waiver or reduction |
The Secretary must grant a waiver or reduction of fees if necessary (1) to protect the public health, (2) if the fee would be a significant barrier to innovation, or (3) if the applicant is a small business that is submitting its first human drug application. In deciding whether to grant a waiver or reduction, the Secretary may consider only the circumstances and assets of the applicant and any affiliate of the applicant. A small business is an entity with fewer than 500 employees, including employees of affiliates, and that does not have a drug product that has been approved under a human drug application and introduced or delivered for introduction into interstate commerce. |
|
(e) Effect of failure to pay fees |
A human drug application or supplement that is not accompanied by the appropriate fee shall be considered incomplete; the Secretary shall not accept it for filing until all fees have been paid. |
|
(f) Limitations |
Fees shall be refunded for a fiscal year unless appropriations for FDA salaries and expenses are equal to or greater than the appropriations (excluding fees) for FY1997, as adjusted for that fiscal year. |
|
(g) Crediting and availability of fees |
Authorized fees may be collected and available for obligation only in the amounts provided in appropriation acts, and may remain available until expended. Fees must be available to defray increases in the cost of resources allocated to the human drug review process. The Secretary may accept early payment of authorized fees. |
|
(h) Collection of unpaid fees |
Any unpaid fee shall be treated as a claim of the United States Government. |
|
(i) Written requests for waivers, reductions, and refunds |
A sponsor must submit a written request to the Secretary for any waiver, reduction, or refund not later than 180 days after the fee is due. |
|
(j) Construction |
"This section may not be construed to require that the number of full-time equivalent positions in the Department of Health and Human Services, for officers, employers, and advisory committees not engaged in the process of the review of human drug applications, be reduced to offset the number of officers, employees, and advisory committees so engaged." |
|
(k) Orphan Drugs |
An orphan drug, as designated under FFDCA Section 526, is exempt from prescription drug program fees under specified conditions if the drug is owned or licensed and is marketed by a company whose previous year gross worldwide revenue was less than $50 million. |
|
FFDCA Sec. 736B [21 U.S.C. 379h—2]. Reauthorization; Reporting Requirements. |
|
|
(a) Performance report |
Each fiscal year for which user fees are collected, the Secretary must submit to Congress a report on FDA's progress in meeting the performance goals specified in the Commitment Letter. FDARA added additional reporting requirements such as "real time reporting" to include specified quarterly posting on the FDA website and inclusion of additional material regarding guidance issued, public meetings held, approvals made, applications filed, rationales for PDUFA program changes, and specified analysis of the meeting of performance goals. |
|
(b) Fiscal report |
Each fiscal year for which user fees are collected, the Secretary must submit to Congress a report on the use by FDA of collected user fees. |
|
(c) Corrective action report |
Each fiscal year for which PDUFA fees are collected, the Secretary must submit a corrective action report to Congress, which includes (1) for PDUFA goals that have been met, recommendations on how the Secretary can improve and streamline the human drug application review process, and (2) for PDUFA goals that have not been met, justification and description of the circumstances under which application review goals were missed and a description of efforts FDA has put in place to improve the ability of the agency to meet such goals. |
|
(d) Enhanced communication |
Each fiscal year, as applicable and requested, representatives from FDA Centers with expertise in human drug review must meet with representatives of the specified congressional committees to report on the contents of the performance, fiscal, and corrective action reports. If requested, FDA representatives must participate in public hearings before the committees regarding the contents of the reports. The hearings must be held not later than 120 days after the end of each fiscal year. |
|
(e) Public Availability |
The Secretary must make the performance and fiscal reports available on the FDA website. |
|
(f) Reauthorization |
The Secretary must, in preparation for the next PDUFA reauthorization, consult with congressional committees, scientific and academic experts, health care professionals, representatives of patient and consumer advocacy groups, and the regulated industry in developing recommendations, including goals and plans for meeting the goals. Prior to beginning negotiations with the regulated industry, the Secretary must request public input via publication of a notice in the Federal Register requesting input, a public meeting, a 30-day comment period, and publication of public comments on the FDA website. At least monthly while negotiating with industry, the Secretary must hold discussions with representatives of patient and consumer advocacy groups. After negotiations with industry, the Secretary must present recommendations to the congressional committees; publish them in the Federal Register; provide 30 days for written comments from the public; hold a public meeting; and, "after consideration of such public views and comments, revise such recommendations as necessary." The Secretary must make publicly available on the FDA website the meeting minutes of all agency negotiations with the regulated industry, including summaries of any substantive proposals made by any negotiating party and any significant controversies or differences of opinions and their resolution. The Secretary must then present the recommendations to Congress, publish the recommendations in the Federal Register, provide for public comment, hold a public meeting, and revise recommendations as necessary. The revised recommendations must be transmitted to Congress by January 15, 2022. |
Source: FFDCA §§735, 736, and 736B.
Notes: CPI = Consumer Price Index, FDA = Food and Drug Administration, FFDCA = Federal Food, Drug and Cosmetic Act, HHS = Health and Human Services, and PDUFA = Prescription Drug User Fee Act
a. Additional dollar amounts, as specified in FFDCA §736(b)(1)(F), are as follows: $20.1 million for FY2018, $21.3 million for FY2019, $17 million for FY2020, $5.4 million for FY2021, and $2.8 million for FY2022.
Appendix C. Abbreviations Used in This Report
|
ARIA |
Active Risk Identification and Analysis |
|
BLA |
Biologics License Application |
|
BsUFA |
Biosimilar User Fee Act |
|
CBER |
Center for Biologics Evaluation and Research |
|
CDER |
Center for Drug Evaluation and Research |
|
CPI |
Consumer Price Index |
|
ESG |
Electronic System Gateway |
|
FDA |
Food and Drug Administration |
|
FDAAA |
Food and Drug Administration Amendments Act |
|
FDAMA |
Food and Drug Administration Modernization Act |
|
FDASIA |
Food and Drug Administration Safety and Innovation Act |
|
FFDCA |
Federal Food, Drug, and Cosmetic Act |
|
FTE |
Full-Time Equivalent |
|
FY |
Fiscal Year |
|
GCP |
Good Clinical Practice |
|
GDUFA |
Generic Drug User Fee Amendments |
|
GLP |
Good Laboratory Practice |
|
GMP |
Good Manufacturing Practice |
|
GRMP |
Good Review Management Principles |
|
HHS |
Department of Health and Human Services |
|
HR |
Human Resources |
|
IND |
Investigational New Drug |
|
IT |
Information Technology |
|
MAPP |
Manual of Policies and Procedures |
|
MIDD |
Model-Informed Drug Development |
|
NDA |
New Drug Application |
|
NME |
New Molecular Entity |
|
OCP |
Office of Combination Products |
|
PD |
Position Description |
|
PDUFA |
Prescription Drug User Fee Act |
|
RDP |
Rare Disease Program |
|
REMS |
Risk Evaluation and Mitigation Strategies |
|
RWE |
Real World Evidence |
|
S. HELP |
Senate Committee on Health, Education, Labor, and Pensions |
|
SOPP |
Standard Operating Policy and Procedure |
|
TAUG |
Therapeutic Area User Guide |
Author Contact Information
Footnotes
| 1. | |
| 2. |
Consolidated Appropriations Act, 2017 (P.L. 115-31). See also CRS Report R44576, The Food and Drug Administration (FDA) Budget: Fact Sheet. |
| 3. |
Ibid. |
| 4. |
Biologics are medical preparations made from living organisms. Examples of such products include traditional biologics (e.g., vaccines, blood, blood products, antitoxins, and allergenics) and human therapeutic agents produced by the biotechnology industry (e.g., insulin, interferon, growth hormone, and epoetin). |
| 5. |
The Prescription Drug User Fee Act (PDUFA), in 1992, added Sections 735 and 736 to the Federal Food, Drug, and Cosmetic Act (FFDCA) [21 U.S.C. 379g and 379h]. In 2007, PDUFA IV added FFDCA Sections 736A and 736B [21 U.S.C. 379h-1 and 379h-2]. |
| 6. |
Philip J. Hilts, "Plan to Speed Approval of Drugs: Makers Would Pay Fees to U.S.," New York Times, August 11, 1992. |
| 7. |
Philip J. Hilts, Protecting America's Health: The FDA, Business, and One Hundred Years of Regulation, New York: Alfred A. Knopf, 2003, p. 278. |
| 8. |
See CRS Report R41983, How FDA Approves Drugs and Regulates Their Safety and Effectiveness. |
| 9. |
FFDCA §735. |
| 10. |
For a description of the FDA drug approval process, see CRS Report R41983, How FDA Approves Drugs and Regulates Their Safety and Effectiveness. |
| 11. |
FFDCA §735(6), as amended by PDUFA I-IV, defines the term "process for the review of human drug applications" as activities necessary for the review of human drug applications and supplements; the issuance of action letters; inspection of prescription drug establishments and other facilities; activities necessary for the review of applications for licensure of biological product establishments and for the release of lots of biologics; monitoring of research conducted in connection with the review of human drug applications; and postmarket safety activities, including adverse event data collection systems and development of analytical tools, and enforcement of study and label-change requirements. PDUFA V and VI did not change this definition. |
| 12. |
FDA, "White Paper Prescription Drug User Fee Act (PDUFA): Adding Resources and Improving Performance in FDA Review of New Drug Applications," November 10, 2005, https://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM149130.pdf. |
| 13. |
Ibid. |
| 14. |
Ibid. |
| 15. |
See also CRS Report R42366, Prescription Drug User Fee Act (PDUFA): 2012 Reauthorization as PDUFA V. |
| 16. |
Under PDUFA I through V, each manufacturer was required to pay an establishment fee—an annual fee for each of its manufacturing establishments—and a product fee—an annual fee for each product that fits within PDUFA's definition. |
| 17. |
PDUFA VI adds the limitation that a person named as the applicant in an approved application cannot be assessed more than five program fees in a fiscal year for prescription drug products identified in such approved application (NDA or BLA). This means that if, for example, there are seven strengths of a drug or biologic approved under one application, the applicant would be charged only five program fees for that application. |
| 18. |
FFDCA §736(b)(2). |
| 19. |
U.S. Congress, House Committee on Energy and Commerce, Subcommittee on Health, Examining FDA's Prescription Drug User Fee Program, 115th Cong., 1st sess., March 22, 2017, http://docs.house.gov/meetings/IF/IF14/20170322/105752/HHRG-115-IF14-Wstate-WoodcockJ-20170322.pdf. |
| 20. |
See CRS Report R42366, Prescription Drug User Fee Act (PDUFA): 2012 Reauthorization as PDUFA V. |
| 21. |
Ibid. |
| 22. |
Under FFDCA §736(c)(1), the inflation adjustment for a fiscal year is to equal the sum of: (1) the average annual percent change in FDA personnel costs (i.e., compensation and benefits) for the first three of the preceding four fiscal years, multiplied by the proportion of personnel compensation and benefits costs to total costs of the process for the review of human drug applications (as defined in FFDCA §735(6)) for the first three years of the preceding four years, and (2) the CPI figure, which is the average annual percent change in the CPI (for DC-MD-VA-WV) for the first three years of the preceding four years of available data multiplied by the proportion of all costs other than personnel compensation and benefits costs to total costs of the process for the review of human drug applications (as defined in FFDCA §735(6)) for the first three years of the preceding four fiscal years. |
| 23. |
FFDCA §736(c)(2). |
| 24. |
FFDCA §736(c)(2)(B)(ii) defines the adjustment percentage as "the weighted change in the 3-year average ending in the most recent year for which data are available, over the 3-year average ending in the previous year, for—(I) the total number of human drug applications, efficacy supplements, and manufacturing supplements submitted to the Secretary; (II) the total number of active commercial investigational new drug applications; and (III) the total number of formal meetings scheduled by the Secretary, and written responses issued by the Secretary in lieu of such formal meetings," as identified in the draft Commitment Letter. |
| 25. |
Under PDUFA V, the final year adjustment provision allowed the Secretary to increase total fee revenue if necessary to provide for up to three months of operating reserves for the process of human drug application review for the first three months following sunset. |
| 26. |
FFDCA §736(c)(3). |
| 27. |
FFDCA §736(c)(4). |
| 28. |
Additional dollar amounts, as specified in FFDCA §736(b)(1)(F), are as follows: $20.1 million for FY2018, $21.3 million for FY2019, $17 million for FY2020, $5.4 million for FY2021, and $2.8 million for FY2022. |
| 29. |
FDA, Priority Review, http://www.fda.gov/ForPatients/Approvals/Fast/ucm405405.htm. |
| 30. |
John K. Jenkins, Director, Office of New Drugs, Center for Drug Evaluation and Research (CDER), FDA. "CDER New Drug Review: 2016 Update," presentation at FDA/CMS Summit, December 14, 2016, slide 4, https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM533192.pdf. Performance goals include both a goal (e.g., review standard NDAs/BLAs in 10 months) and a performance measure (e.g., review in 10 months for 90% of standard NDAs/BLAs). The Agreement sets a goal for each type of submission. |
| 31. |
For additional information about the other medical product user fees, see CRS Report R44750, FDA Medical Product User Fee Reauthorization: In Brief. |
| 32. |
FDA, PDUFA VI: Fiscal Years 2018-2022, https://www.fda.gov/forindustry/userfees/prescriptiondruguserfee/ucm446608.htm. |
| 33. |
81 Federal Register 46929, July 19, 2016. |
| 34. |
CRS Report R44961, FDA Reauthorization Act of 2017 (FDARA, P.L. 115-52). |