On December 31, 2019, the World Health Organization (WHO) was informed of a cluster of pneumonia cases in Wuhan City, Hubei Province of China. Illnesses have since been linked to a disease caused by a previously unidentified strain of coronavirus, designated Coronavirus Disease 2019, or COVID-19. Despite containment efforts in China, the United States, and elsewhere, by late February there were indications that the COVID-19 outbreak may have entered a new phase, with community spread occurring or suspected in several countries other than China, including in the United States.1
Diagnostic testing is a critical part of the public health response to and clinical management of the COVID-19 caused by the SARS-CoV-2 virus. Efforts in the United States to develop and disseminate a test for COVID-19 have faced challenges. Manufacturing and quality issues with the nation's test—developed by the Centers for Disease Control and Prevention (CDC)—resulted in essentially all testing going through CDC's laboratory facility in Atlanta through early March, despite distribution of test kits to state and local public health labs beginning in early February. CDC's initial test kits had to be remanufactured and redistributed, which, along with other factors, has significantly delayed access to testing throughout the country. It has been reported that the CDC's Atlanta laboratory is currently under investigation by the Department of Health and Human Services (HHS) for possible quality issues related to its manufacture of the test kits, which may have led to the contamination of one reagent and thus to the quality issues with the test.2
In this context, on February 29, 2020, in an effort to facilitate the expansion of testing capacity as the first cases of community spread were confirmed in the United States, the Food and Drug Administration (FDA) announced a significant new policy. This policy, issued via agency guidance and effective immediately, allows certain laboratories—principally clinical and commercial laboratories—that have developed and validated their own COVID-19 diagnostics to begin to use the tests prior to the test receiving an Emergency Use Authorization (EUA) from the agency.3
Diagnostic testing for COVID-19, in part because it is caused by a novel pathogen, has been led and carried out to date through the country's public health infrastructure. This includes primarily the CDC and the country's network of state and local public health laboratories. In contrast, in most situations, diagnostic testing is carried out by a number of facilities, including private commercial laboratories (e.g., Quest, LabCorp), hospital and other clinical laboratories, and laboratories in academic medical centers, among others.
FDA's February 29 guidance aims to facilitate the expansion of diagnostic testing from the public health setting into the clinical health care and commercial settings, leveraging significant standing resources throughout the country, including facilities, trained personnel, expertise, materials, and equipment. It is FDA's intention that this expansion will help the country meet the increasing and substantial demand for testing generated by community spread of the disease and expanded clinical testing guidelines issued by the CDC. In addition, because many cases of COVID-19 are reportedly mild or asymptomatic, widespread access to testing—which informs development of important metrics such as the case fatality rate—is critical to understanding the scope and extent of the disease in the United States, and to efficiently directing resources to mitigate its impact in the broader community.
Diagnostic tests—formally called in vitro diagnostic (IVD) devices—may be commercially developed and distributed as "kits" or developed, validated, and carried out by a single laboratory. This second type of test—known as a laboratory-developed test, or an LDT—is the more commonly used type of test because of its flexibility and differing federal regulatory requirements, among other reasons. Although the CDC's test is being manufactured as a test kit and initially has been distributed to specific CDC-qualified labs, the FDA guidance targets LDTs that are generally carried out in clinical and academic settings or private commercial laboratories. All clinical laboratories in the United States, regardless of whether they are part of the country's public health infrastructure or part of the health care delivery system, are regulated by the Clinical Laboratory Improvement Amendments of 1988 (CLIA) program, administered by the Centers for Medicare & Medicaid Services (CMS).
Federal agencies involved in the regulation of IVDs include FDA and CMS. FDA derives its authority to regulate the sale and distribution of medical devices, such as IVDs, from the Federal Food, Drug, and Cosmetics Act (FFDCA) and the Public Health Service Act (PHSA). CMS's authority to regulate IVDs is through CLIA (P.L. 100-578). FDA regulates the safety and effectiveness of diagnostic tests, as well as the quality of the design and manufacture of the diagnostic test. CMS regulates the quality of clinical laboratories and the clinical testing process.
Diagnostic Tests
What Are IVD Tests?
In vitro diagnostic devices are defined in FDA regulation as a specific subset of medical devices that include "reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions ... in order to cure, mitigate, treat, or prevent disease ... [s]uch products are intended for use in the collection, preparation, and examination of specimens taken from the human body." As indicated by this definition, an IVD may also include components of tests, which can include both non-diagnostic ingredients, called general purpose reagents (GPRs), and the active ingredient in a diagnostic test, referred to as the analyte specific reagent (ASR).
In general, an IVD device may be a "commercial test kit" (a product developed, produced, and sold by a manufacturer for distribution to multiple laboratories) or a "laboratory developed test" (a product developed by and used in a single laboratory). LDTs may use components (e.g., general purpose reagents like a buffer) that are either manufactured in-house by the laboratory or commercially developed and distributed.
What Is an LDT?
A laboratory-developed test is a class of IVD that is designed, manufactured, and used within a single laboratory. LDTs are often used to test for conditions or diseases that are either rapidly changing (e.g., new strains of known infectious diseases) or the subject of quickly advancing scientific research (e.g., genomic testing for cancer). The majority of genetic tests—a type of IVD that analyzes various aspects of an individual's genetic material (e.g., DNA, RNA)—are LDTs.
How Are IVD Tests Regulated?
In general, oversight of in vitro diagnostic devices focuses on ensuring their safety and effectiveness; their accuracy and reliability; the quality of clinical laboratories that carry out IVD testing; the utility of the information generated by IVDs in clinician and patient decision-making; and the truthfulness of claims made about IVDs that are marketed directly to consumers. As with other medical devices, the application of FDA regulatory requirements to IVDs depends on the IVD's risk classification according to its intended use. Classification is based, in turn, on the risk the device poses to the patient. For IVDs, which are informational tests, the risk to the patient is that of an incorrect test result, either a false positive or a false negative result, either of which may cause serious harm to the individual. In the case of infectious diseases—for example, COVID-19—the risk of a false negative test extends beyond the individual patient into the community at large. The FDA has three classes of medical device: Class I (low risk), Class II (moderate risk), and Class III (high risk). Regulatory controls are dependent on the class of a given medical device. COVID-19 diagnostics would most likely fall in Class III, as they would be considered to be high-risk devices.
How Are LDTs Regulated?
The regulation of laboratory-developed tests has been the subject of ongoing debate over the past 20 years, driven in large part by an increase in the number and complexity of genetic tests over this time. In general, the FDA has maintained that it has clear regulatory authority over LDTs, as it does with all IVDs that meet the definition of medical device in the FFDCA.4 However, the FDA traditionally exercised enforcement discretion over LDTs—choosing not to enforce applicable statutory and regulatory requirements with respect to such tests—meaning that most of these tests have neither undergone premarket review nor received FDA clearance or approval for marketing. To date, FDA has instead focused its enforcement efforts on commercial IVD kits, which are broadly commercially marketed. In recent years, despite the absence of specific agency guidance on the regulation of LDTs, FDA has nevertheless begun to assert authority over LDTs, and specifically over some direct-to-consumer (DTC) genetic tests, that it considers to be higher-risk tests.
What Is CLIA and How Is It Involved in LDT Regulation?
CLIA of 1988 provides CMS with authority to regulate clinical laboratories.5 CLIA establishes quality standards for clinical laboratory testing and a certification program for clinical laboratories that perform testing using IVD devices. All laboratories that perform diagnostic testing for health-related reasons (i.e., with results returned to the patient or a health care practitioner) are regulated by CMS under the authority of CLIA. For CLIA to apply, testing must be carried out on a human specimen. CLIA certification is based on the level of complexity of testing that the laboratory performs, specifically (1) low (therefore, waived) complexity, (2) moderate complexity, and (3) high complexity. FDA is responsible for categorizing tests according to their level of complexity. CLIA requirements are used to evaluate a test's analytical validity, defined as the ability of a test to detect or measure the analyte it is intended to detect or measure. Laboratories that perform moderate- and high-complexity testing must meet specific standards and requirements as a condition of certification, including proficiency testing (PT), patient test management, quality control, personnel qualifications, and quality assurance. All LDTs, including genetic tests offered as LDTs, are considered high-complexity tests under CLIA. All COVID-19 diagnostics would be considered to be high complexity tests under CLIA.
How Are IVDs Regulated by the FDA During an Emergency Such as the Outbreak of COVID-19?
In certain public health or other emergency situations, the HHS Secretary may declare that existing circumstances justify the use of unapproved medical products for certain uses, or approved medical products for unapproved uses.6 This declaration facilitates access to not-yet-approved medical products in an expedited manner during certain emergency situations. In the case of the COVID-19 disease, HHS Secretary Azar determined that "there is a public health emergency and [declared] that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for detection and/or diagnosis of the novel coronavirus."7 On the basis of this declaration, FDA issued an Emergency Use Authorization authorizing the emergency use of the CDC-developed diagnostic test for COVID-19.8 The FDA also issued an EUA to the state of New York for an LDT developed by the state public health labs. To date, these are the only two EUAs for coronavirus diagnostics that the FDA has issued.9
How Does the Emergency Use Authority Apply to LDTs if They Are Generally Exempted from Premarket Requirements?
During an emergency, all laboratory-developed tests testing for the relevant pathogen (in this case, SARS-CoV-2) must either be approved, cleared, or authorized under an EUA to be legally carried out. As noted above, FDA generally waives most regulatory requirements (e.g., premarket review) for LDTs in normal situations; nevertheless, LDTs may only be used with authorization (e.g., an EUA) during an emergency declaration pursuant to FFDCA Section 564.10 That is, statutory requirements under FFDCA Section 564 apply to LDTs as they do to other medical products, and they apply to both commercial test kits—which are normally subject to FDA regulatory requirements—and to LDTs.
The EUA process is usually used to expedite access to medical products that would otherwise need premarket approval or clearance in emergency situations. However, because premarket approval requirements for LDTs are generally waived through enforcement discretion by the agency, the EUA represents additional regulatory requirements for the use of an LDT in emergency situations. This is because, among other things, in the case of a communicable disease, the test result has implications beyond the individual being tested, and so a false negative result could have serious consequences for the community.11 Therefore, FDA states that these tests need an EUA in an emergency prior to clinical use as do other medical products. In contrast, for commercial test kits, the EUA represents an abbreviated mechanism that allows the unapproved product to be used without undergoing the FDA premarket review typically required (for a complex molecular test for a novel pathogen, that review would generally be a Premarket Approval, or PMA, for high-risk medical devices).
Despite a request from the Association of Public Health Laboratories (APHL) to FDA, the agency declined to exercise enforcement discretion with respect to LDTs that detect SARC-CoV-2 and diagnose COVID-19 and the requirement that they receive an EUA prior to use. APHL maintains that these labs are regulated by CLIA, and that this regulatory oversight is sufficient.12 However, on February 29, 2020, FDA announced a new policy allowing certain laboratories that have developed and validated COVID-19 LDTs to begin to use the test clinically prior to it receiving an EUA from the agency but after validation of the test and notification of the agency (see "What Steps Did FDA Take to Expand Testing Capacity in Response to the Problems with CDC's Test?").13
The CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time Reverse Transcriptase (RT)-PCR Diagnostic Panel
How Does the CDC's COVID-19 Diagnostic Test Work?
The diagnostic test developed by the CDC, called the 2019-Novel Coronavirus (2019-nCoV) Real-Time Reverse Transcriptase (RT)-PCR Diagnostic Panel, is a complex molecular diagnostic test that relies on generally standard molecular biology laboratory techniques. Specifically, the test uses a technique called Polymerase Chain Reaction (PCR), a standard in vitro technique for amplification of DNA. Because the SARS-CoV-2 virus—the virus that causes COVID-19—is an RNA14 virus, the RNA must be first reverse transcribed to generate copy DNA, or cDNA, which is then amplified (multiple copies are generated) using PCR. PCR relies on primers—very short single stranded pieces of DNA that are complementary to and bind with specific regions of the viral genome and thus define the specific genomic region to be amplified. The test then relies on a probe, or a single-stranded piece of DNA that is chemically or radioactively labelled, that can bind to and thus detect the amplified target portion of the viral genetic material.
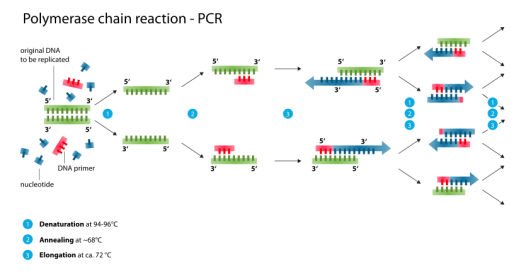 |
|
Source: https://upload.wikimedia.org/wikipedia/commons/thumb/9/96/Polymerase_chain_reaction.svg/835px-Polymerase_chain_reaction.svg.png, accessed March 3, 2020. |
{kind=link}
CDC's original test used three sets of primers and probes: two to target specific regions of a designated gene within the SARS-CoV-2 viral genome, and a third that was specific to all SARS-like coronaviruses (see "What Quality Problems Did the CDC's Test Experience on Rollout to the State and Local Public Health Laboratories?"). The test also includes a number of authorized control samples, including a positive control for SARS-CoV-2 and a "no template control" to test for system contamination. Together, these controls help ensure that the test is functioning properly.
What Type of IVD Is the CDC's Test and Who May Carry It Out?
The CDC's test is being developed by the agency as a test kit and is generally authorized to be distributed to state and local public health laboratories to augment public health testing capacity. The test received an EUA from the FDA on February 4, 2020, under which "authorized laboratories" may receive and carry out the test despite the fact that it is not FDA-approved or FDA-cleared, and that it does not meet all related regulatory requirements for marketing.15 The EUA notes that "[t]esting is limited to qualified laboratories designated by CDC and, in the United States, certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), (42 U.S.C. §263a), to perform high complexity tests."16 CDC-qualified laboratories with a CLIA certification for high-complexity testing able to receive the test kits include U.S. state and local public health laboratories, Department of Defense (DOD) laboratories, and select international laboratories. The public health laboratories must verify the test themselves prior to using it, and are currently required to send presumptive positive cases back to the CDC in Atlanta for confirmatory testing by the agency.
What Is the Role of the Commercial Manufacturer IDT in the Development and Distribution of the CDC's Test Kit?
The Trump Administration had estimated that approximately 1 million tests would be broadly available imminently, facilitated by leveraging manufacturing of the CDC test kit by a private company, Integrated DNA Technologies (IDT), a manufacturer that has been working with CDC.17 According to HHS Secretary Azar on March 5, 2020, IDT will send the CDC test kit to "hospitals, other labs around the country, commercial, public health … labs" by the end of the week of March 2.18 The test kit being manufactured by IDT, identical to the CDC test kit and referred to as "IDT 2019-novel coronavirus kit," is being qualified by CDC in lots, and FDA reports that these CDC-qualified, IDT-manufactured test kits are covered by the CDC's EUA authorization of February 4, 2020.19
FDA notes that if a lab purchases a test kit from IDT, the laboratory does not need its own EUA to carry it out, but that "[t]esting using CDC's EUA-authorized protocol and CDC qualified lots of reagents is considered to be testing done under the CDC's EUA. Labs performing such testing should follow any applicable conditions set forth in the EUA."20 LabCorp, a large commercial laboratory, is already reporting that it can perform the CDC test "if needed to meet testing demand" and that the test is only for use with "authorized specimens collected from individuals who meet CDC criteria for COVID-19 testing."21 IDT is manufacturing test kits in two sizes, with the largest having a capacity of 500 reactions (approximately 250 individual patients).22
What Quality Problems Did the CDC's Test Experience on Rollout to the State and Local Public Health Laboratories?
As noted above, the CDC's test kit used three sets of probes and primers—or reagents—to detect and identify viral DNA beyond that specific to COVID-19. One of these reagents, the one meant to detect any SARS-like coronavirus including SARS-CoV-2, was returning inconclusive results. In response, the CDC validated a new protocol for their test that allows it to be run excluding the faulty reagent, running the test with only the other two diagnostic components. CDC has the authority to modify the test through enforcement discretion granted by FDA.23 The agency determined that the exclusion of this reagent does not affect the accuracy or the sensitivity and specificity of the test.
Certain laboratories have continued to experience problems running the test, even when using the modified protocol, with at least one laboratory reporting that the first reagent was also returning inconclusive results. This problem severely limited the state and local public health laboratories' ability to carry out the CDC's test.
In response to these issues, the New York State Department of Health requested and was granted the FDA's second EUA for its own laboratory-developed test, the New York SARS-CoV-2 Real-time RT-PCR Diagnostic Panel.24 Testing is limited under the EUA to two laboratories in New York—the Wadsworth Center, New York State Department of Public Health, and the New York City Department of Health and Mental Hygiene, Public Health Laboratories. New York was one of the states that had continued difficulty implementing CDC's original test kit, even with the modified protocol.
In response, CDC is also manufacturing new test kits after reportedly resolving the manufacturing issue that affected the original test kit. This time, however, CDC is manufacturing test kits with only the two reagents that were unaffected by the quality issue—that are specific to SARS-CoV-2—and those kits are being made available to qualified CDC labs through the International Reagent Resource.25,26 These test kits are expected to result in the public health laboratories having capacity to test up to 75,000 patients.27
Some believe that the CDC's choice to develop and mitigate quality problems with its own COVID-19 diagnostic when an accepted diagnostic was available through the World Health Organization (WHO), which was efficiently distributing a German-developed test globally early in the outbreak, was a decision that cost the United States time in its response to the virus's introduction and spread in the country.28 Some speculated about the use of the third reagent—that was to detect SARS-like coronaviruses—and whether it had been strictly necessary if the test was still accurate at diagnosing COVID-19 without that reagent included in the test at all, or if it had instead overcomplicated the test.29 In general, there have been questions raised about the CDC's handling of the development and distribution of its test, and its response to the quality problems that occurred, and the impact this may have had on the country's ability to detect community spread of the disease before it occurred more widely.30
What Steps Did FDA Take to Expand Testing Capacity in Response to the Problems with CDC's Test?
On February 29, 2020, as problems with the rollout of the CDC-developed diagnostic test continued, FDA announced a new policy to immediately leverage LDTs developed in high-complexity commercial, reference, and clinical laboratories nationwide to expand testing capacity. Specifically, the new agency guidance allows CLIA-certified high-complexity laboratories that have developed and validated their own COVID-19 diagnostics to use the tests while the laboratory is preparing, and FDA is reviewing, their EUA submission.31,32
The FDA guidance states that laboratories have 15 days after validating their test to submit an EUA application to FDA, and the guidance recommends confirming the test's first five negative and positive results against an EUA-authorized diagnostic. According to FDA, it "does not intend to object to the use of these tests for clinical testing while the laboratories are pursuing an EUA with the FDA. Importantly, this policy only applies to laboratories that are certified to perform high-complexity testing consistent with requirements under Clinical Laboratory Improvement Amendments."33 The guidance includes detailed information about FDA's expected methods for test validation.
Pursuant to this FDA guidance, on March 5, 2020, LabCorp announced that it had begun offering its LDT, the LabCorp 2019 Novel Coronavirus (COVID-19), NAA test "for ordering by physicians or other authorized healthcare providers anywhere in the U.S.," and that it is currently pursuing an EUA with the agency for the test.34 This test is to take between three to four days to return results, and a health care provider must order and authorize it, and obtain the required specimen from the patient. Under the FDA's new guidance, Quest has also announced that it will begin testing for coronavirus with its own LDT, beginning March 9, 2020, and that it plans to pursue an EUA with the agency within 15 days, as required by the guidance.35